
独家原创 | 盛荣副教授:靶向核受体的抗前列腺癌小分子药物研发新进展
转自:药学进展

盛荣

浙江大学副教授,浙江省新药审评专家,浙江省杰出青年基金获得者,浙江省“151”人才,《中国现代应用药学》编委,浙江省药学会药化和抗生素专委会委员。盛荣副教授针对肿瘤、病毒、炎性疾病等重大疾病,将计算机辅助药物设计(CADD)、蛋白降解靶向嵌合体(PROTAC)等前沿技术与经典药物设计相融合,开展基于靶点的新药研发和转化研究,发现临床候选分子(PCC),并开展高端仿制药的非专利绿色工艺、新晶型、新盐型研究,协助企业报批生产。近年来以第一、通信作者在JMedChem,BriefBioinform,OrgLett等期刊共发表SCI论文70余篇,获得授权美国专利1项,中国发明专利23项。作为主要研制人员研发的APG-1387和BIOS-0618分别进入Ⅱ期和Ⅰ期临床研究;主持国家自然科学基金5项,国家新药创制重大专项、国家支撑计划、省科技厅领雁项目、省科技厅重点项目、省自然科学基金重点、省自然杰出青年基金等,研究成果荣获浙江省科技进步二等奖4项,三等奖1项。
靶向核受体的抗前列腺癌小分子药物研发新进展 PPS
袁乐儿1#,廖金标1#,蔡吕涛1,2,胡陈娴1,杨号东2,盛荣1,2*
(1浙江大学药学院,浙江杭州310058;2.浙江大学金华研究院,浙江金华321002)
[摘要]作为全球男性第二大常见癌症,前列腺癌严重威胁着人类健康。在前列腺癌发病过程中,雄激素受体(androgenreceptor,AR)起着关键作用,糖皮质激素受体(glucocorticoidreceptor,GR)也参与调节部分AR通路下游基因,因此,阻断AR和GR信号通路是前列腺癌治疗的重要策略。综述从核受体AR和GR的结构特征和生物学功能出发,从药物化学角度系统介绍了近5年来靶向核受体的抗前列腺癌药物研发新进展,包括雄激素竞争性AR拮抗剂、雄激素非竞争性AR拮抗剂、AR降解剂、GR拮抗剂和AR/GR双重拮抗剂等,为去势抵抗性前列腺癌的治疗提供新思路。
2022年全球癌症统计资料显示,前列腺癌(prostatecancer,PCa)已成为男性发病率第2的癌症,致死率排名第5[1]。前列腺癌发病机制复杂,影响因素众多。研究表明,核受体家族蛋白在前列腺癌的发生发展中具有重要作用,核受体主要有雄激素受体(androgenreceptor,AR)、糖皮质激素受体(glucocorticoidreceptor,GR)和盐皮质激素受体(mineralocorticoidreceptor,MR)等。其中,AR信号通路在前列腺癌发生和发展过程中起关键作用[2],拮抗AR通路是治疗激素敏感性前列腺癌及去势抵抗性前列腺癌(castration-resistantprostatecancer,CRPC)的重要策略。目前,第2代AR拮抗剂恩杂鲁胺已成为一线治疗药物,但随着AR拮抗剂的广泛使用,临床耐药已经产生,耐药机制包括AR基因扩增、AR配体结合域上的点突变和缺乏配体结合域(ligand-bindingdomain,LBD)的剪接变体(ARvariants,ARVs)产生[3]。
临床研究发现,CRPC的耐药往往伴随着GR表达的上调。由于AR和GR高度相似,在CRPC中,GR可绕过AR阻断,代替AR调控AR蛋白的表达,促进肿瘤细胞增殖[4]。研究表明,GR拮抗剂可恢复前列腺癌细胞对AR拮抗剂的敏感性,因此,同时阻断AR和GR信号通路成为克服CRPC耐药性的有效策略[5]。
本文对近5年(2018—2023)靶向核受体AR和GR的抗前列腺癌药物研发新进展进行综述,包括雄激素竞争性AR拮抗剂、雄激素非竞争性AR拮抗剂、AR降解剂、GR拮抗剂和AR/GR双重拮抗剂,旨在为CRPC的治疗提供新思路。
1
雄激素受体
1.1雄激素受体的结构及功能
AR是由919个氨基酸组成的类固醇受体转录因子,为核受体家族成员。AR基因位于染色体Xq12上,由8个外显子组成,如图1A所示。AR由N端结构域(N-terminaldomain,NTD)、DNA结合域(DNA-bindingdomain,DBD)、铰链区(hinge,H)和LBD组成。外显子1编码的NTD包含激活功能区1(activationfunction1,AF1),支持AR的转录活性。外显子2和3编码的DBD包含2个锌指结构,第1个锌指结构决定DNA结合特异性,第2个锌指结构与AR二聚化及DNA受体复合物的稳定性有关。C端包含由外显子4编码的柔性铰链区和外显子5~8编码的高度保守的LBD,LBD包含配体结合口袋(ligand-bindingpocket,LBP)、激活功能区2(activationfunction2,AF2)和结合功能区3(bindingfunction3,BF3)[6]。
1.2雄激素受体的信号通路
AR的信号通路如图1B所示,未与雄激素[睾酮或二氢睾酮(dihydrotestosterone,DHT)]结合的AR蛋白位于细胞质中,与热休克蛋白(heatshockprotein,HSP)形成稳定的复合物。雄激素与AR的LBD中的LBP结合后,诱导AR构象发生变化,随后,AR与HSP分离,发生同源二聚化,并易位至细胞核内,二聚化AR的DBD与DNA上的雄激素应答元件(androgenresponseelement,ARE)结合后,招募一系列转录共调节因子,进而调节近百种AR靶基因的表达,包括前列腺特异性抗原(prostate-specificantigen,PSA)、跨膜丝氨酸蛋白酶2(transmembraneproteaseserine2,TMPRSS2)等,该过程的过度激活促进前列腺癌的进展[7]。
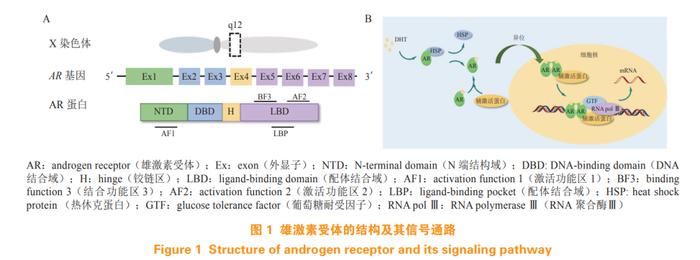
2
雄激素竞争性雄激素受体拮抗剂
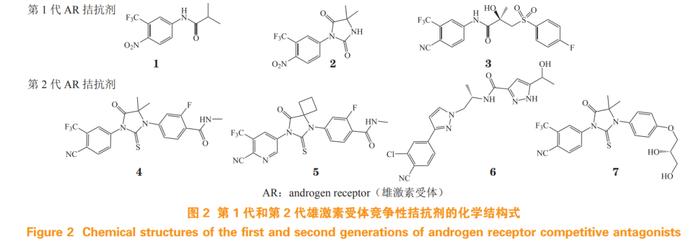
AR的激活高度依赖于雄激素与LBP的结合,因此靶向AR的LBP拮抗雄激素的作用,是近年AR拮抗剂新药研发的主要策略。目前已上市的第1代和第2代AR拮抗剂(结构式见图2)均结合于ARLBP位点,其作用机制为LBP与雄激素竞争性结合从而阻断AR激活,进而抑制AR信号通路,降低血清PSA水平[6]。
2.1第1代竞争性雄激素受体拮抗剂
氟他胺(flutamide,1)于1989年获美国食品药品监督管理局(FoodandDrugAdministration,FDA)批准用于治疗PCa,是首个上市非甾体类AR拮抗剂。通过进一步的结构优化得到的尼鲁他胺(nilutamide,2)和比卡鲁胺(bicalutamide,3),分别于1995年和1996年获批。第1代AR拮抗剂与AR的亲和力较弱,不能充分阻断AR通路,与雄激素剥夺疗法(androgen-deprivationtherapy,ADT)联用可适度延长患者总生存期,但不良反应发生率较高,且长期用药易引起ARLBD的耐药突变,包括ARW742C/L和H875Y/T878A突变等[6],导致拮抗剂转变为部分激动剂,使患者产生耐药性,最终发展为CRPC[8]。
2.2第2代竞争性雄激素受体拮抗剂
恩杂鲁胺(enzalutamide,4)是首个第2代AR拮抗剂,与AR结合亲和力显著提升,能抑制AR核易位、与雄激素结合以及与靶基因结合的全过程,分别于2012年、2018年和2019年被FDA批准用于治疗转移性去势抵抗性前列腺癌(metastaticcastration-resistantprostatecancer,mCRPC)、非转移性去势抵抗性前列腺癌(non-metastaticcastration-resistantprostatecancer,nmCRPC)和激素敏感性前列腺癌(hormone-sensitiveprostatecancer,HSPC)[9];但是因在患者脑内恩杂鲁胺稳态水平较高,导致拮抗γ-氨基丁酸α(γ-aminobutyricacidα,GABAα)受体从而易产生癫痫等不良反应。阿帕鲁胺(apalutamide,5)为恩杂鲁胺的类似物,该药对AR的拮抗活性更高,分别于2018年和2019年获批用于治疗nmCRPC和mHSPC[9],且不易入脑,中枢神经系统(centralnervoussystem,CNS)不良反应较少。但ARLBD的F877L突变使恩杂鲁胺和阿帕鲁胺转变为激动剂,以及ARVs的产生也导致耐药[10]。
达洛鲁胺(darolutamide,6)由拜耳和奥利安公司联合研发,该药能有效抑制恩杂鲁胺和阿帕鲁胺耐药的ARF877L突变体,延长高危nmCRPC患者的无转移生存期,于2019年获FDA批准用于治疗nmCRPC[11]。但是患者发生疲劳、疼痛等副作用的比例较高,导致临床应用受限[12]。
恒瑞医药研发的瑞维鲁胺(rezvilutamide,7)也为恩杂鲁胺类似物,在mCRPC的Ⅰ/Ⅱ期临床试验中,该药160mg·d-1剂量与360mg·d-1恩杂鲁胺的药物暴露量相当,且耐受性良好[最大耐受剂量(maximaltolerancedose,MTD)大于480mg·d-1];同时,该药还具有血脑屏障透过率低、诱发癫痫风险小和安全性高等优点[13]。瑞维鲁胺联合ADT治疗高瘤负荷mHSPC,可显著延长患者总生存期,于2022年6月获国家药品监督管理局(NationalMedicalProductsAdministration,NMPA)批准治疗mHSPC[14]。
目前,还有多种AR拮抗剂处于临床试验阶段(结构式见图3)。例如,HC-1119(8)为氘代恩杂鲁胺,针对mCRPC患者的Ⅰ期临床试验显示,与恩杂鲁胺相比,HC-1119代谢缓慢、半衰期更长,母体药物的血浆浓度增加约40%,主要代谢物的血浆浓度降低约75%[15],目前正开展治疗mCRPC上市申请。
开拓药业的普克鲁胺(9)与AR的结合亲和力比恩杂鲁胺高3.4倍[半数抑制浓度(halfmaximalinhibitoryconcentration,IC50)为32nmol·L-1,抑制常数(inhibitionconstant,Ki)为14nmol·L-1)],且对ARF877L,ARW742C/L和ARH875Y/T878A等耐药突变体有效。该药在体内消除速度慢,单次和连续给药的平均表观清除率分别为0.55~1.30和0.17~0.46L·h-1[16],且CNS分布低,不易诱发癫痫,mCRPC的Ⅱ期临床试验效果良好[17]。
西安杨森的TRC-253(10)可强效抑制野生型AR和F877L突变体,IC50分别为54和37nmol·L-1,在F877L突变体的异种移植模型中抑瘤效果显著[18],正处于mCRPCⅡ期临床[19]。
目前,正大天晴的TQB3720[20]、日本大鹏药品的TAS-3681[21]、康朴的X-Synergy®(结构均尚未披露)处于Ⅰ期临床研究。
此外,还有多种AR拮抗剂正处于临床前研究阶段(结构式见图3)。其中,化合物11在LNCaP细胞中的AR转录抑制活性与恩杂鲁胺相近[8],化合物12在LNCaP细胞中的活性较恩杂鲁胺提高近60倍[22]。(+)JJ-450(13)经高通量筛选和结构优化得到[23],在C4-2-PSA-rl细胞中,其活性与恩杂鲁胺相当,作用机制为延缓AR核易位,且对恩杂鲁胺耐药的CRPC有效,能抑制剪接变体AR-V7的转录活性和基因表达[24]。苯甲酰衍生物14和15,对表达AR野生型(wild-type,WT)、T877A和H874Y突变体的前列腺癌细胞,均具有显著的增殖抑制作用[25]。
侯廷军团队基于ARLBD结构,经虚拟筛选发现AT2(16)为全新骨架AR拮抗剂,其抗AR转录活性的IC50为0.06µmol·L-1,能抑制AR下游靶基因及AR核易位[26]。该团队采用分子动力学模拟构造ARLBD二聚体结构,并进行虚拟筛选和结构优化,其中香豆素类衍生物17抑制AR转录的IC50为0.17µmol·L-1,机制可能是抑制AR二聚化[27]。
以达洛鲁胺为先导化合物,对其2-氯苯腈部分和吡唑部分进行结构改造,其中,类似物18能有效拮抗AR转录活性(IC50=0.05µmol·L-1),对ARF876L和ART877A的效力均优于达洛鲁胺,且抑制AR下游靶基因表达[28]。
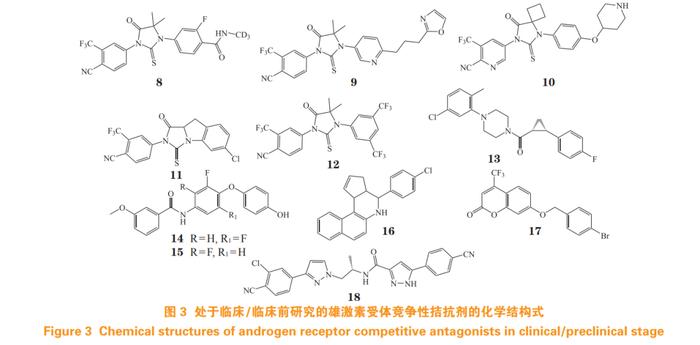
3
非雄激素竞争性雄激素受体拮抗剂
ARLBP易突变的性质严重限制经典AR竞争性拮抗剂的临床使用。近年来,靶向AR雄激素非竞争性位点的拮抗剂也得到了发展,如靶向LBD的AF2和BF3,NTD的AF1以及DBD的拮抗剂(结构式见图4),以寻求克服耐药的前列腺癌新疗法。
3.1靶向配体结合域激活功能区2的拮抗剂
AF2是AR与雄激素结合发生构象变化后在LBD表面形成的疏水口袋,在该位点招募共调节因子对AR发挥转录活性至关重要。AF2可通过与共调节因子LXXLL和FXXLF基序特异性相互作用来募集辅助激活因子。AF2作为转录激活结构域,在正常细胞中调节功能较弱,而在CRPCAR高表达的环境中趋于主导。因此,靶向AF2直接阻断AR与辅助激活因子相互作用的疗法,理论上不会受LBP耐药突变的影响,从而具有良好的临床应用前景[29]。
针对AF2蛋白的虚拟筛选而发现的苯磺酰胺类化合物IMB-A6(19),可有效抑制WT-AR和AR突变体(ART877A和ARF876L)的转录活性。免疫共沉淀实验证明,IMB-A6靶向AF2位点,破坏AR与共调节因子脯氨酸-谷氨酸-亮氨酸富集蛋白1(proline-,glutamicacid-andleucine-richprotein1,PELP1)在AF2位点的相互作用,从而抑制AR活性[30]。
双苯甲酰胺类衍生物20的AR转录抑制活性优异,IC50为16nmol·L-1,且100nmol·L-1浓度下能有效抑制LNCaP细胞活性,并抑制AR调控基因PSA的表达。机制研究表明,该化合物在AF2位点抑制AR与共调节因子PELP1的相互作用,从而发挥AR拮抗活性[31]。
基于AF2蛋白进行虚拟筛选发现的磺酰胺类化合物T1-12(21),其AR转录抑制活性优良,IC50为0.47μmol·L-1,能显著抑制AR下游基因表达和AR核易位,在LNCaP异种移植模型的效果与恩杂鲁胺相当。经时间分辨荧光共振能量转移技术(time-resolvedfluorescenceresonanceenergytransfer,TR-FRET)AR共激活因子试剂盒验证,T1-12能浓度依赖性地下调FRET信号,通过阻断AF2与共调节因子FXXLF结合,从而发挥AR拮抗活性[32]。
3.2靶向配体结合域结合功能区3的拮抗剂
BF3位点在空间上与AF2相邻,BF3通过招募FK506结合蛋白52(FK506bindingprotein52,FKBP52)和Bcl-2结合抗凋亡蛋白1(Bcl-2-associatedathanogene-1,BAG-1)等辅助激活因子来调控AR转录活性。同时,BF3还与相邻的AF2形成变构网络以调节ARLBD功能。前列腺癌和雄激素不敏感综合征中大量的BF3突变(前者如Gln670等;后者如Leu830,Pro723等),以及BF3抑制剂氟灭酸的发现,证明靶向BF3作为治疗前列腺癌策略的可行性[33]。
Leblanc等[34]发现2-(1H-吲哚-3-基)喹啉衍生物VPC-13566(22)具有良好的AR转录抑制活性(IC50=0.06μmol·L-1),但体内代谢不稳定,半衰期短(T1/2=21min)。对吲哚及喹啉环进行修饰后得到的衍生物VPC-13789(23),代谢稳定性显著提高(T1/2=206min),能有效抑制WT-AR(IC50=0.19μmol·L-1)和ARF877L突变体(IC50=0.21μmol·L-1)的活性,抑制LNCaP细胞的增殖。将VPC-13789制备为可口服的磷酸钠前药VPC-13822(24),VPC-13822在CRPC异种移植模型中疗效良好,显著降低血清PSA水平,且长期毒性小,具有明显的临床价值。
Chen等[35]基于恩杂鲁胺和达洛鲁胺化学结构,经骨架跃迁设计得到一系列衍生物,其中化合物25的AR转录抑制活性优良(IC50=0.07μmol·L-1),对ARF877L/T878A双突变体有效(IC50=0.25μmol·L-1),且能有效抑制LNCaP细胞增殖(IC50=6.23μmol·L-1),在小鼠LNCaP异种移植瘤模型中,经口给药(100mg·kg-1·d-1)28天后,可有效抑制肿瘤生长,分子动力学模拟结果预测该化合物可能结合于BF3位点。
3.3靶向配体结合域二聚体界面口袋的拮抗剂
侯廷军团队通过分子动力学模拟和小角度X射线散射实验,发现AR的二聚体界面口袋(dimerinterfacepocket,DIP),并通过基于结构的虚拟筛选,发现小分子拮抗剂M17-B15(26)能够有效地破坏AR蛋白二聚化,进而抑制AR信号转导。其AR转录抑制活性优良(IC50=0.03μmol·L-1),且对恩杂鲁胺耐药的ARF876L/T877A双突变体有效(IC50=0.15μmol·L-1),能有效抑制AR调控及基因转录和翻译水平。在异种移植LNCaP细胞模型中,瘤内注射M17-B15(2.5mg·kg-1·d-1)可显著抑制肿瘤生长。因此,靶向DIP位点是全新的AR拮抗剂研发策略[36]。
3.4靶向N端结构域的拮抗剂
NTD是AR完全转录活性的关键区域,并存在于所有形式的AR中,且NTD可以在雄激素非依赖性前列腺癌细胞中调节AR活性[21]。因此,靶向NTD的拮抗剂有望解决CRPC耐药性问题。
从海绵提取物库中筛选而得的EPI-001的最有效的立体异构体为EPI-002(27),该化合物能与NTD上的TAU-5结合[37],其乙酰基前药EPI-506(28)为首个进入临床的NTD拮抗剂,但因药物剂量负担过重、口服生物利用度差而终止试验[38]。类似物EPI-7386(29)在LNCaP异种移植模型中活性与恩杂鲁胺相当;在恩杂鲁胺耐药的VCaP异种移植模型中,EPI-7386单药或与恩杂鲁胺联用,均表现出显著的抗肿瘤活性,目前处于mCRPC的Ⅰ/Ⅱ期临床试验[39]。
通过靶向ARNTD的虚拟筛选获得的QW07(30),具有良好的AR转录抑制活性(IC50=4.93μmol·L-1),能有效抑制LNCaP和22RV1细胞增殖。在CRPC动物模型中,QW07(10mg·kg-1·d-1)显著抑制22RV1和VCaP肿瘤的生长。染色质免疫沉淀实验证明,QW07与ARNTD的结合抑制AR转录复合物的形成,从而阻止下游基因与启动子、增强子的结合[40]。
研究发现DHT可诱导AR入核,并发生液-液相分离,从而形成激活的转录凝集体,该过程主要由NTD驱动,采用ARF877L/T878A细胞株对化合物库进行筛选,获得化合物ET0516(31),并经微尺度热电泳等实验证明该化合物结合在ARNTD。ET0516可有效地抑制野生型和耐药突变的AR的相分离形成,在5.0μmol·L-1浓度下,对LNCaP和VCaP细胞增殖抑制率大于50%[41]。
3.5靶向DNA结合域的拮抗剂
DBD是AR与AREs结合不可或缺的结构域,对AR-FL和雄激素非依赖ARVs的核定位至关重要。因此,靶向DBD的拮抗剂可以直接阻断AR与DNA的相互作用,以克服CRPC耐药性[42]。
基于DBD结构虚拟筛选发现的噻唑吗啉衍生物VPC-14449(32)具有优良的AR转录抑制活性(IC50=0.34μmol·L-1),能有效抑制LNCaP细胞和恩杂鲁胺耐药MR49F细胞的增殖,生物膜干涉实验结果证明,该化合物选择性地靶向ARDBD-铰链区结构域表面,进而阻断AR与DNA相互作用[43]。
二氢查尔酮衍生物MF-15(33)为AR和AKR1C3的双重抑制剂。AKR1C3为参与雄激素生物合成的酶,与恩杂鲁胺耐药CRPC有关。在10µmol·L-1浓度下,MF-15对AKR1C3的抑制率为87%,并能浓度依赖性地抑制AR-FL和AR-V7的活性,显著抑制AR下游PSA表达。此外,MF-15抑制DBD中的P-box相互作用而发挥AR拮抗作用[44]。
基于DBD晶体的虚拟筛选发现了苯甲酸衍生物Cpd39(34),其AR转录抑制活性中等(IC50=10.94μmol·L-1),能减少WT-AR和AR-V7调控基因的表达,且抑制AR下游PSA基因表达,生物膜干涉实验结果表明,该化合物靶向DBD-ARE结合界面位点,抑制ARDBD-DNA相互作用[45]。
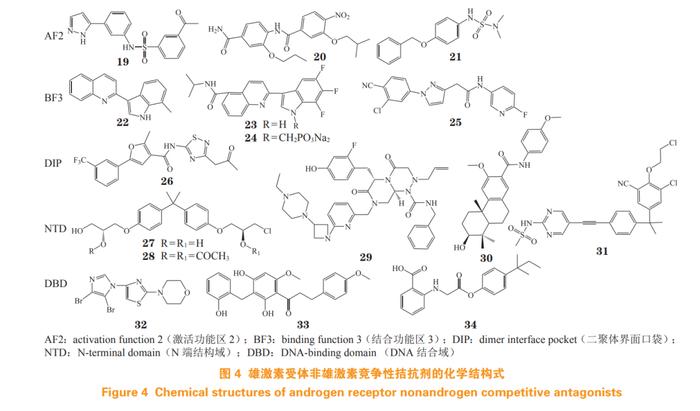
4
雄激素受体降解剂
4.1选择性雄激素受体降解剂
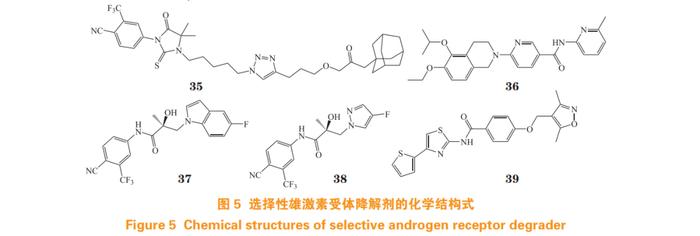
临床患者对AR拮抗剂耐药的快速出现,使得对靶向AR的新策略需求增加,其中选择性雄激素受体降解剂(selectiveandrogenreceptordegrader,SARD)是克服耐药的策略之一(结构式见图5)[46]。目前,SARD的确切作用机制尚不明确,一般认为是通过泛素-蛋白酶体系统(ubiquitin-proteasomesystem,UPS)介导AR的降解,可能是通过增强AR与E3连接酶——双微体同源基因2(murinedoublemimute2,MDM2)的关联而发挥作用。以靶向LBP的AR拮抗剂RU59063母核作为AR配体,并通过烷基链和三氮唑连接疏水标签(hydrophobictag,HyT),得到化合物A9(35),其AR转录抑制活性良好(IC50=1.75μmol·L-1),使用10μmol·L-1A9处理LNCaP细胞72h后,可基本实现AR的完全降解[47]。
经虚拟筛选发现的二氢异喹啉烟酰胺衍生物EIQPN(36),其作用位点为AF1,在10μmol·L-1浓度下,其AR抑制率大于95%,对LNCaP细胞增殖抑制率大于90%。在NTD过表达HEK293T细胞中,EIQPN的IC50为0.78μmol·L-1。作用机制研究表明,EIQPN能有效降低LNCaP,CWR22rv,DU145,PPC1和HEK293T细胞的AR和ARVs水平,且能抑制异种移植小鼠模型CWR22rv肿瘤生长[48]。
对AR拮抗剂比卡鲁胺采用合环策略,得到类似物UT-155(37)和UT-34(38)。作用机制研究表明,两者均能与AF1位点结合,并具有SARDs功能。体外活性测试表明,在1.0μmol·L-1浓度下,两者在LNCaP和22RV1细胞系中均能有效降解全长AR和ARVs,并对恩杂鲁胺耐药MR49F细胞生长抑制率大于70%。在100mg·kg-1剂量下,UT-155和UT-34在LNCaP去势模型和MR49F模型中均能有效抑制肿瘤生长[49]。
Wu等[50]经药效团虚拟筛选发现了小分子SARDZ15(39),靶点验证发现该化合物为AF1与LBD双位点SARD。体外活性测试表明,Z15在LNCaP细胞中可明显下调AR蛋白表达水平,半数最大降解浓度(thehalf-maximaldegradationconcentration,DC50)为1.05μmol·L-1。5μmol·L-1的Z15和环己酰胺(100μg·mL-1)联用处理LNCaP细胞24h后,可基本实现AR完全降解。在22Rv1细胞中,Z15对AR和AR-V7的DC50分别为1.16和2.24μmol·L-1。
4.2雄激素受体蛋白降解靶向嵌合体
作为一种新兴的靶蛋白降解技术,蛋白降解靶向嵌合体(proteolysistargetingchimera,PROTAC)近年来迅速发展,引起越来越多的关注。PROTAC由3个部分组成:与E3泛素连接酶结合的配体,与靶蛋白(proteinofinterst,POI)结合的配体以及连接2个配体的连接链。PROTAC分子与E3泛素连接酶和POI形成三元复合物,特异性诱导POI的泛素化标记,进而通过泛素-蛋白酶体途径降解多泛素化的靶蛋白[51]。
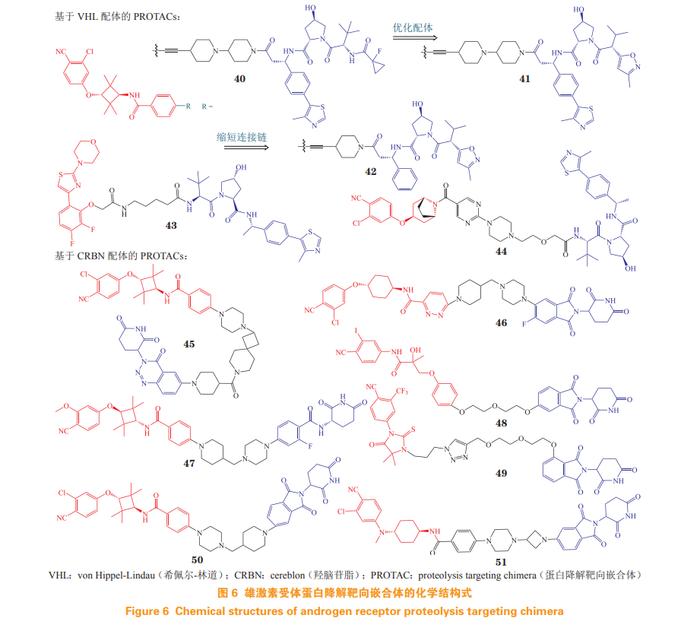
第1个靶向AR的PROTAC分子是含有E3泛素连接酶的肽基配体,其理化性质和细胞通透性较差,限制了进一步的应用。2008年,Crews等[52]首次报道以MDM2抑制剂为E3连接酶配体,比卡鲁安作为AR配体的小分子ARPROTAC,该化合物能在微摩尔浓度下降解AR蛋白,从而开启小分子ARPROTAC药物(结构式见图6)时代。
4.2.1基于希佩尔-林道E3泛素连接酶配体的蛋白降解靶向嵌合体芳氧基环丁基胺衍生物是高活性AR拮抗剂[53],其骨架常作为AR配体,以希佩尔-林道(vonHippel-Lindau,VHL)配体为E3泛素连接酶配体,通过刚性接头将两者连接得到PROTAC分子ARD-69(40)[54]。对ARD-69的活性进行评估,实验结果表明,ARD-69在LNCaP,VCaP和22Rv1细胞中的DC50分别为0.86,0.76和10.4nmol·L-1。细胞增殖抑制实验中,该化合物对LNCaP,VCaP和22Rv1细胞的IC50分别为0.25,0.34和183nmol·L-1,活性为恩杂鲁胺的100倍以上。腹腔给药50mg·kg-1ARD-69后,在48h内有效减少VCaP异种移植小鼠的AR蛋白和PSA蛋白。ARD-61(41)是优化VHL配体得到的新PROTAC分子[55],表现出良好的体内外AR降解活性,且对恩杂鲁胺耐药模型以及AR阳性乳腺癌有效。
对ARD-61进一步进行结构修饰,将不同AR配体与VHL配体进行组合得到ARD-266(42)[56]。ARD-266在1nmol·L-1浓度下即可诱导LNCaP,VCaP和22Rv1细胞的AR蛋白降解,与ARD-61相比,ARD-266在保持良好的AR降解效率和前列腺癌拮抗活性的同时,具有更小的相对分子质量,且理化性质和成药性更佳。
MTX-23(43)由DBD配体、柔性连接链和VHL配体组成[57],由于该化合物结合于DBD,因此对AR-FL和AR-V7具有显著降解活性,DC50分别为2.00和0.37µmol·L-1,它能有效抑制CRPC细胞增殖,经口给药(8.3mg·kg-1)显著抑制恩杂鲁胺耐药的前列腺癌异种移植瘤(如22Rv1肿瘤),且MTX-23对AR-V7的降解效果优于AR-FL,其深入的机制仍在探索中。
化合物A031(44)由具有芳氧基托品环的独特AR配体、VHL配体和包含哌嗪和嘧啶环的连接体组成。在VCaP异种移植斑马鱼中,8.3μmol·L-1的化合物A031表现出55%的肿瘤生长抑制率,与恩杂鲁胺相当,药代动力学性质良好,毒性较低[58]。
4.2.2基于羟脑苷脂配体的蛋白降解靶向嵌合体Takwale等[59]通过使用芳氧基环丁胺AR拮抗剂和羟脑苷脂(cereblon,CRBN)配体得到TD-802(45),该化合物是首个报道的具有CRBN配体的ARPROTAC。TD-802在LNCaP细胞中的DC50为12.5nmol·L-1,在体内异种移植物小鼠模型中能有效抑制肿瘤生长,并表现出良好的肝微粒体稳定性和体内药代动力学性质。
ARV-110(46)由阿维纳斯公司研发,经过对恩杂鲁胺和多种CRBN配体及多种连接链的组合筛选,及进一步结构优化而得,为最早进入临床的ARPROTAC分子[60]。在体内对恩杂鲁胺获得性和内在耐药模型中,经口给予3.0mg·kg-1ARV-110,分别显示出70%和100%的肿瘤生长抑制率。ARV-110在Ⅰ期临床试验中用于mCRPC患者,具有良好的耐受性、安全性和药代动力学性质[61]。2022年2月,阿维纳斯公司披露ARV-110在治疗mCRPC的临床试验中,具有持续抗肿瘤活性和患者获益的证据,在携带ART878X/H875Y(T878X为T878A或T878S)突变肿瘤患者中,ARV-110使46%患者的PSA水平降低50%以上。目前,ARV-110正在开展Ⅱ期临床试验[62]。
目前,另有多个ARPROTAC已进入临床试验,其中阿维纳斯公司的ARV-766(47),相对于ARV-110,不仅对H875Y和T878A等多种突变体亚型具有降解能力,而且能更有效地降解与阿比特龙和其他AR途径拮抗剂耐药相关的L702H突变体亚型,并在动物模型中得以验证[63]。2021年9月,ARV-766在美国开展Ⅱ期临床试验,用于治疗mCRPC;其结构在2023年4月的美国癌症研究协会(AmericanAssociationforCancerResearch,AACR)年会中被披露。此外,百时美施贵宝公司研发的CC-94676、冰洲石生物科技的AC-0176以及海创药业研发的HP518等PROTAC分子都处于Ⅰ期临床试验阶段,用于治疗mCRPC[64]。
Kim等[65]以比卡鲁胺为AR配体,通过柔性链与CRBN配体连接,设计并合成了一系列ARPROTAC。其中化合物48能以剂量和时间依赖性的方式降解AR蛋白(LNCaP:DC50=5.21µmol·L-1)。另一系列的ARPROTAC以恩杂鲁胺衍生物为AR配体,通过不同的三氮唑片段连接CRBN配体,其中化合物49具有优良的AR结合亲和力(85%)和AR降解活性[66]。
Han等[67]以芳氧基环丁胺为AR配体,采用含哌嗪的连接链与CRBN配体沙利度胺相连,分别得到ARD-2128(50)和ARD-2585(51)。ARD-2128在VCaP和LNCaP细胞中的DC50分别为0.28和8.3nmol·L-1,能抑制AR调控基因,经口给药可有效降低肿瘤组织中AR蛋白,有效抑制小鼠肿瘤生长,且毒性较低[68]。ARD-2585的降解活性更高,在VCap和LNCaP细胞中的DC50均低于0.10nmol·L-1。ARD-2585比恩杂鲁胺可更有效地降解全长AR和ARVs,经口生物利用度(小鼠)达51%,体内疗效优于恩杂鲁胺,颇具临床应用潜力。
ARPROTAC为CRPC的治疗提供了新的治疗策略,但ARPROTAC的相对分子质量较大,成药性亟需提升,实现良好的口服生物利用度具有挑战性。此外,ARPROTAC多数作用于LBP口袋,对于缺乏LBD的ARVs无效。因此,后续的ARPROTAC的研究方向将聚焦解决上述问题。
5
糖皮质激素受体及其拮抗剂的应用
5.1糖皮质激素受体的结构与功能
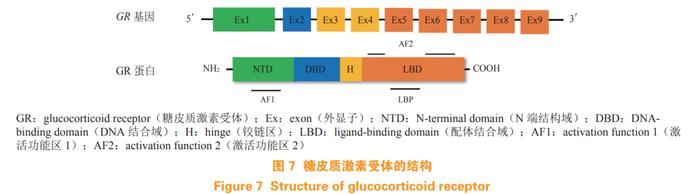
人源GR基因位于5号染色体上,由9个外显子组成。如图7所示,GR主要包含4个区域,NTD域由外显子2编码,该区域主要负责转录激活功能以及和辅助调节因子结合;DBD域由外显子3和4编码,其结构上包含了2个锌指结构识别DNA上的糖皮质激素反应元件(glucocorticoid-responsiveelements,GREs);外显子5~9编码铰链区(H)和LBD域,LBD域包含1个配体结合口袋(LBP),以及1个AF2结构域,以配体依赖的方式与辅助调节因子相互作用[69]。
5.2糖皮质激素受体与去势抵抗性前列腺癌
在前列腺癌中,AR与TLE3(transducin-likeenhancerofsplit3)会和GR的增强子相结合;另外,多梳抑制复合物2(polycombrepressivecomplex2,PRC2)会导致组蛋白3上的第27位赖氨酸的三甲基化(trimethylationoflysine27onhistone3,H3K27me3),并通过zeste基因增强子同源物2(enhancerofzestehomolog2,EZH2)沉积到GR启动子和增强子上,两者共同抑制GR的表达[70]。在早期前列腺癌中,GR表现为下调趋势,且在AR激活的情况下,GR可能发挥抑制肿瘤发生发展的作用。而在CRPC患者中,恩杂鲁胺等AR拮抗剂的使用,使得AR的表达受到抑制,并且在治疗过程中TLE3会出现表达缺失,进而上调GR增强子上的H3K27乙酰化,使原先H3K27沉积受到抑制,从而恢复GR的表达[70]。而GR表达升高后,会与ARE结合,共同调控一些经典的AR靶基因,导致前列腺癌的生长和恩杂鲁胺耐药的发生。另外,有研究表明GR介导的葡萄糖转运蛋白4(glucosetransporter4,GLUT4)上调与前列腺癌治疗中出现的恩杂鲁胺耐药性和交叉耐药性相关,抑制GR或GLUT4后,可以减少葡萄糖的摄取,改善癌细胞的耐药性[71]。目前研究认为,GR和AR既存在协同作用,也存在对抗作用,其作用与肿瘤进展关系密切[72]。综上,GR具有成为克服CRPC耐药治疗关键靶点的潜力,具有器官靶向性的GR拮抗剂及GR/AR双重拮抗剂(结构式见图8)可能在治疗中更具有优势。
5.3针对糖皮质激素受体过表达的治疗
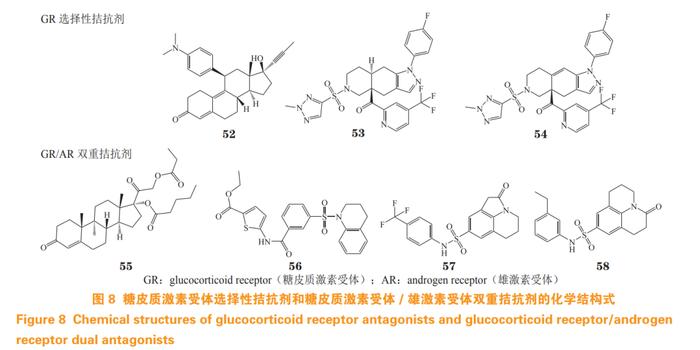
5.3.1联合糖皮质激素受体拮抗剂用药治疗去势抵抗性前列腺癌研究发现GR拮抗剂可恢复耐药细胞对恩杂鲁胺的敏感性,从而提出联合用药治疗因GR过表达引起的CRPC耐药。2013年,米非司酮(mifepristone,52)和恩杂鲁胺联用治疗CRPC的临床试验(临床试验编号:NCT02012296)启动,但因疗效不明显而终止[72]。2018年,已开展GR选择性拮抗剂exicorilant(53)和relacorilant(54)分别与恩杂鲁胺联用治疗CRPC的临床试验(临床试验编号:NCT03437941,NCT03674814),但目前临床数据尚未公布。
5.3.2雄激素受体/糖皮质激素受体双重拮抗剂 联用方式治疗因GR过表达而引起的CRPC耐药具有一定的意义,但药物-药物相互作用的存在使临床应用受限。因此,靶向AR/GR的双重拮抗剂是治疗GR过表达CRPC的新方向。
对脱氧可的松结构改造所得的化合物CB-03-10(55)能有效抑制AR和GR的转录活性,对LNCaP细胞和恩杂鲁胺耐药LNCaP-EnzaR细胞均有效,并能抑制AR/GR下游靶基因和蛋白的表达。在小鼠异种移植LNCaP细胞模型中,CB-03-10的活性与紫杉醇相当,具有良好的开发潜力[73]。
基于ARLBP蛋白虚拟筛选发现的化合物Z19(56)对AR和GR均具有良好的转录抑制活性,IC50分别为2.03和2.50μmol·L-1。Z19能抑制表达AR突变22Rv1细胞系和AR阴性PC-3细胞系生长,降低GR和AR信号下游蛋白和mRNA的水平,有效抑制CRPC耐药肿瘤的增殖。生物膜干涉实验和分子对接研究表明,该化合物可能靶向AR和GR的LBP口袋,与内源性配体竞争性结合AR/GR而发挥拮抗作用[74]。
基于ARLBP蛋白虚拟筛选发现的化合物H18(57)具有一定的AR拮抗活性,经分子动力学模拟研究及结构优化得到AR/GR双重拮抗剂HD57(58),该化合物对AR和GR转录抑制IC50分别为0.39和17.81μmol·L-1。此外,该分子对大部分AR突变体的抑制活性与达洛鲁胺相当,并能抑制AR下游靶基因PSA的表达和AR核易位[75]。
6
结语与展望
AR和GR与前列腺癌的发生、发展密切相关,是抗前列腺癌新药研发的重要靶标。不过,目前已上市的AR拮抗剂均靶向AR的LBP位点,且存在交叉耐药性,限制了临床上的使用。因此,靶向AR的非LBP位点引起越来越多的关注,包括ARLBD上的AF2和BF3位点,AR的DIP口袋,AR的NTD及DBD域等,尤其是后2个靶点,有望解决ARVs缺乏LBD域的问题。另一方面,近年来ARPROTAC已成为AR靶向治疗的热点,ARV-110和ARV-766已分别进入Ⅱ期临床试验,且疗效明显,前景光明。另外,基于ARDBD抑制剂的PROTAC52535455565758(如MTX-23),为治疗AR-V7导致的CRPC亚群提供了新的解决方案。GR拮抗剂以及AR/GR双重拮抗剂具有独特的作用机制,在CRPC治疗领域预期有更广阔的前景。
综上所述,靶向核受体AR和GR是临床治疗前列腺癌的重要策略,虽然耐药突变、PROTAC生物利用度低等问题亟需解决,但相信随着计算机辅助药物设计(computeraideddrugdesign,CADD)、人工智能(artificialintelligence,AI)和PROTAC技术的进一步发展,将会有更多的靶向核受体抗前列腺癌新药进入临床,为广大患者提供更好的治疗选择。
参考文献:
[1]SiegelRL,MillerKD,WagleNS,etal.Cancerstatistics,2023[J].CACancerJClin,2023,73(1):17-48.
[2]HeY,XuW,XiaoY,etal.Targetingsignalingpathwaysinprostatecancer:mechanismsandclinicaltrials[J].SignalTransductTargetTher,2022,7(7):2337-2367.
[3]SchmidtKT,HuitemaADR,ChauCH,etal.Resistancetosecondgenerationandrogenreceptorantagonistsinprostatecancer[J].NatRevUrol,2021,18(4):209-226.
[4]SmithR,LiuMQ,LibyT,etal.Enzalutamideresponseinapanelofprostatecancercelllinesrevealsaroleforglucocorticoidreceptorinenzalutamideresistantdisease[J].SciRep,2020,10(1):1-13.
[5]AroraVK,SchenkeinE,MuraliR,etal.Glucocorticoidreceptorconfersresistancetoantiandrogensbybypassingandrogenreceptorblockade[J].Cell,2013,155(6):1309-1322.
[6]WestabyD,DeLaMazaMDF,PaschalisA,etal.Anewoldtarget:androgenreceptorsignalingandadvancedprostatecancer[J/OL].AnnuRevPharmacolToxicol,2022,62:131-153[2023-06-12].https://pubmed.ncbi.nlm.nih.gov/34449248/.DOI:10.1146/annurevpharmtox-052220-015912.
[7]XiangWG,WangSM.Therapeuticstrategiestotargettheandrogenreceptor[J].JMedChem,2022,65(13):8772-8797.
[8]KonoM,FujiiT,LimB,etal.Androgenreceptorfunctionandandrogenreceptor-targetedtherapiesinbreastcancerareview[J].JAMAOncol,2017,3(9):1266-1273.
[9]ChenY,ZhouQ,HankeyW,etal.Secondgenerationandrogenreceptorantagonistsandchallengesinprostatecancertreatment[J/OL].CellDeathDis,2022,13(7):632[2023-06-12].https://doi.org/10.1038/s41419-022-05084-1.
[10]KorpalM,KornJM,GaoX,etal.AnF876LmutationinandrogenreceptorconfersgeneticandphenotypicresistancetoMDV3100(enzalutamide)[J].CancerDiscov,2013,3(9):1030-1043.
[11]MoilanenAM,RiikonenR,OksalaR,etal.DiscoveryofODM-201,anew-generationandrogenreceptorinhibitortargetingresistancemechanismstoandrogensignaling-directedprostatecancertherapies[J/OL].SciRep,2015,5(1):12007[2023-06-12].https://doi.org/10.1038/srep12007.
[12]RizzoA,MerlerS,SorgentoniG,etal.Riskofcardiovasculartoxicitiesandhypertensioninnonmetastaticcastration-resistantprostatecancerpatientstreatedwithnovelhormonalagents:asystematicreviewandmeta-analysis[J].ExpertOpinDrugMetabToxicol,2021,17(10):1237-1243.
[13]QinX,JiD,GuW,etal.ActivityandsafetyofSHR3680,anovelantiandrogen,inpatientswithmetastaticcastration-resistantprostatecancer:aphaseI/IItrial[J/OL].BMCMed,2022,20(1):84[2023-06-12].https://doi.org/10.1186/s12916-022-02263-x.
[14]GuW,HanW,LuoH,etal.Rezvilutamideversusbicalutamideincombinationwithandrogen-deprivationtherapyinpatientswithhighvolume,metastatic,hormone-sensitiveprostatecancer(CHART):arandomised,open-label,phase3trial[J].LancetOncol,2022,23(10):1249-1260.
[15]MaH,XuW,NiJ,etal.PhaseIclinicaltrialofHC-1119softcapsuleinChinesehealthyadultmalesubjects:pharmacokineticsandsafetyofsingle-doseproportionalityandeffectsoffood[J].Prostate,2022,82(2):276-285.
[16]ZhouT,XuW,ZhangW,etal.PreclinicalprofileandphaseIclinicaltrialofanovelandrogenreceptorantagonistGT0918incastrationresistantprostatecancer[J/OL].EurJCancer,2020,134:29-40[2023-06-12].https://doi.org/10.1016/j.ejca.2020.04.013.
[17]ZhouT,QinS,XuW,etal.Proxalutamideinmetastaticcastrationresistantprostatecancer:primaryanalysisofamulticenter,randomized,open-label,phase2trial[J/OL].IntJCancer,2023[2023-06-12].https://doi.org/10.1002/ijc.34512.
[18]ZhangZ,ConnollyPJ,LimHK,etal.DiscoveryofJNJ-63576253:aclinicalstageandrogenreceptorantagonistforF877Lmutantandwild-typecastration-resistantprostatecancer(mCRPC)[J].JMedChem,2021,64(2):909-924.
[19]RathkopfDE,SalehMN,TsaiFYC,etal.Anopenlabelphase1/2Astudytoevaluatethesafety,pharmacokinetics,pharmacodynamics,andpreliminaryefficacyofTRC253,anandrogenreceptorantagonist,inpatientswithmetastaticcastrationresistantprostatecancer[J/OL].JClinOncol,2019:e16542-e16542[2023-06-12].https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.e16542.
[20]ZhangZ,XieT,ZhangS,etal.Secondgenerationandrogenreceptorantagonist,TQB3720abrogatesprostatecancergrowthviaAR/GPX4axisactivatedferroptosis[J/OL].FrontPharmacol,2023,14:1110146[2023-06-12].https://doi.org/10.3389/fphar.2023.1110146.
[21]SobhaniN,NeeliPK,D’angeloA,etal.AR-V7inmetastaticprostatecancer:astrategybeyondredemption[J/OL].IntJMolSci,2021,22(11):5515[2023-06-12].https://doi.org/10.3390/ijms22115515.
[22]BassettoM,FerlaS,PertusatiF,etal.Designandsynthesisofnovelbicalutamideandenzalutamidederivativesasantiproliferativeagentsforthetreatmentofprostatecancer[J/OL].EurJMedChem,2016,118:230-243[2023-06-12].https://doi.org/10.1016/j.ejmech.2016.04.052.
[23]JohnsonJK,SkodaEM,ZhouJ,etal.Smallmoleculeantagonistsofthenuclearandrogenreceptorforthetreatmentofcastration-resistantprostatecancer[J].ACSMedChemLett,2016,7(8):785-790.
[24]YangZ,WangD,JohnsonJK,etal.Anovelsmallmoleculetargetsandrogenreceptoranditssplicevariantsincastration-resistantprostatecancer[J].MolCancerTher,2020,19(1):75-88.
[25]KazuiY,FujiiS,YamadaA,etal.Structure-activityrelationshipofnovel(benzoylaminophenoxy)phenolderivativesasanti-prostatecanceragents[J].BioorgMedChem,2018,26(18):5118-5127.
[26]TangQ,FuW,ZhangM,etal.Novelandrogenreceptorantagonistidentifiedbystructure-basedvirtualscreening,structuraloptimization,andbiologicalevaluation[J/OL].EurJMedChem,2020,192:112156[2023-06-12].https://doi.org/10.1016/j.ejmech.2020.112156.
[27]FuW,ZhangM,LiaoJ,etal.Discoveryofanovelandrogenreceptorantagonistmanifestingevidencetodisruptthedimerizationoftheligand-bindingdomainviaattenuatingthehydrogen-bondingnetworkbetweenthetwomonomers[J].JMedChem,2021,64(23):17221-17238.
[28]XuQ,ZhangZ,HuangC,etal.Developmentofnovelandrogenreceptorantagonistsbasedonthestructureofdarolutamide[J].BioorgChem,2022,124:105829[2023-06-12].https://doi.org/10.1016/j.bioorg.2022.105829.
[29]SadarMD.Discoveryofdrugsthatdirectlytargettheintrinsicallydisorderedregionoftheandrogenreceptor[J].ExpertOpinDrugDiscov,2020,15(5):551-560.
[30]LiuY,WuM,WangT,etal.Structuralbasedscreeningofantiandrogentargetingactivationfunction-2bindingsite[J/OL].FrontPharmacol,2018,9:1419[2023-06-12].https://doi.org/10.3389/fphar.2018.01419.
[31]LeeTK,RavindranathanP,SonavaneR,etal.Astructure-activityrelationshipstudyofbis-benzamidesasinhibitorsofandrogenreceptor-coactivatorinteraction[J/OL].Molecules,2019,24(15):2783[2023-06-12].https://doi.org/10.3390/molecules24152783.
[32]ChaiX,SunHY,ZhouWF,etal.DiscoveryofN-(4-(benzyloxy)-phenyl)-sulfonamidederivativesasnovelantagonistsofthehumanandrogenreceptortargetingtheactivationfunction2[J].JMedChem,2022,65(3):2507-2521.
[33]KongXT,XingEM,ZhuangT,etal.Mechanisticinsightsintotheallostericinhibitionofandrogenreceptorsbybindingfunction3antagonistsfromanintegratedmolecularmodelingstudy[J].JChemInfModel,2021,61(7):3477-3494.
[34]LeblancE,BanFQ,CavgaAD,etal.Developmentof2-(5,6,7-trifluoro-1H-indol-3-yl)-quinoline-5-carboxamideasapotent,selective,andorallyavailableinhibitorofhumanandrogenreceptortargetingitsbindingfunction-3forthetreatmentofcastration-resistantprostatecancer[J].JMedChem,2021,64(20):14968-14982.
[35]ChenCW,ChaiX,HuXP,etal.Discoveryof2-(1-(3-chloro-4-cyanophenyl)-1H-pyrazol-3-yl)acetamidesaspotent,selective,andorallyavailableantagoniststargetingtheandrogenreceptor[J].JMedChem,2022,65(19):13074-13093.
[36]FuWT,YangH,HuCX,etal.Small-moleculeinhibitionofandrogenreceptordimerizationasastrategyagainstprostatecancer[J].AcsCentSci,2023,9(4):675-684.
[37]DeMolE,FenwickRB,PhangCTW,etal.EPI-001,acompoundactiveagainstcastration-resistantprostatecancer,targetstransactivationunit5oftheandrogenreceptor[J].AcsChemBiol,2016,11(9):2499-2505.
[38]LeMoigneR,ZhouHJ,ObstJK,etal.LessonslearnedfromthemetastaticcastrationresistantprostatecancerphaseItrialofEPI506,afirst-generationandrogenreceptorN-terminaldomaininhibitor[J/OL].JClinOncol,2019[2023-06-12]https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.7_suppl.257.
[39]LeMoigneR,PearsonP,LauriaultV,etal.PreclinicalandclinicalpharmacologyofEPI-7386,anandrogenreceptorN-terminaldomaininhibitorforcastration-resistantprostatecancer[J/OL].JClinOncol,2021,39(6):119[2023-06-12]https://ascopubs.org/doi/10.1200/JCO.2021.39.6_suppl.119.
[40]PengSH,WangJ,ChenH,etal.Regressionofcastration-resistantprostatecancerbyanovelcompoundQW07targetingandrogenreceptorN-terminaldomain[J].CellBiolToxicol,2020,36(5):399-416.
[41]XieJJ,HeH,KongWN,etal.Targetingandrogenreceptorphaseseparationtoovercomeantiandrogenresistance[J].NatChemBiol,2022,18(12):1341-1350.
[42]LiF,SongC,ZhangY,etal.Structuraloverviewandperspectivesofthenuclearreceptors,amajorfamilyasthedirecttargetsforsmall-moleculedrugs[J/OL].ActaBiochimBiophysSin,2022,54:1-13[2023-06-12].https://pubmed.ncbi.nlm.nih.gov/35130630/.DOI:10.3724/abbs.2021001.
[43]DalalK,BanFQ,LiHF,etal.Selectivelytargetingthedimerizationinterfaceofhumanandrogenreceptorwithsmall-moleculestotreatcastration-resistantprostatecancer[J/OL].CancerLett,2018,437:35-43[2023-06-12].https://pubmed.ncbi.nlm.nih.gov/30165195/.DOI:10.1016/j.canlet.2018.08.016.
[44]KafkaM,MayrF,TemmlV,etal.DualinhibitoryactionofanovelAKR1C3inhibitoronbothfull-lengthARandthevariantAR-V7inenzalutamideresistantmetastaticcastrationresistantprostatecancer[J/OL].Cancers,2020,12(8):2092[2023-06-12].https://doi.org/10.3390/cancers12082092.
[45]PangJP,ShenC,ZhouWF,etal.DiscoveryofnovelantagoniststargetingtheDNAbindingdomainofandrogenreceptorbyintegrateddocking-basedvirtualscreeningandbioassays[J].ActaPharmacolSin,2022,43(1):229-239.
[46]GombosA.Selectiveoestrogenreceptordegradersinbreastcancer:areviewandperspectives[J].CurrOpinOncol,2019,31(5):424-429.
[47]XieH,LiangJJ,WangYL,etal.Thedesign,synthesisandantitumormechanismstudyofnewandrogenreceptordegrader[J/OL].EurJMedChem,2020,204:112512[2023-06-12].https://doi.org/10.1016/j.ejmech.2020.112512.
[48]TranTT,SongCH,KimKJ,etal.AnewcompoundtargetstheAF-1ofandrogenreceptoranddecreasesitsactivityandproteinlevelsinprostatecancercells[J].AmJCancerRes,2020,10(12):4607-4623.
[49]HeYL,HwangDJ,PonnusamyS,etal.ExplorationandbiologicalevaluationofbasicheteromonocyclicpropanamidederivativesasSARDsforthetreatmentofenzalutamide-resistantprostatecancer[J].JMedChem,2021,64(15):11045-11062.
[50]WuM,ZhangR,ZhangZ,etal.Selectiveandrogenreceptordegrader(SARD)toovercomeantiandrogenresistanceincastration-resistantprostatecancer[J/OL].Elife,2023,12:e70700[2023-06-12].https://doi.org/10.7554/eLife.70700.
[51]LiD,YuD,LiY,etal.AbibliometricanalysisofPROTACfrom2001to2021[J/OL].EurJMedChem,2022,244:114838[2023-06-12].https://doi.org/10.1016/j.ejmech.2022.114838.
[52]SchneeklothAR,PucheaultM,TaeHS,etal.Targetedintracellularproteindegradationinducedbyasmallmolecule:enroutetochemicalproteomics[J].BioorgMedChemLett,2008,18(22):5904-5908.
[53]GuoC,LintonA,KephartS,etal.Discoveryofaryloxytetramethylcyclobutanesasnovelandrogenreceptorantagonists[J].JMedChem,2011,54(21):7693-7704.
[54]HanX,WangC,QinC,etal.DiscoveryofARD-69asahighlypotentproteolysistargetingchimera(PROTAC)degraderofandrogenreceptor(AR)forthetreatmentofprostatecancer[J].JMedChem,2019,62(2):941-964.
[55]KregelS,WangC,HanX,etal.Androgenreceptordegradersovercomecommonresistancemechanismsdevelopedduringprostatecancertreatment[J].Neoplasia,2020,22(2):111-119.
[56]HanX,ZhaoL,XiangW,etal.DiscoveryofhighlypotentandefficientPROTACdegradersofandrogenreceptor(AR)byemployingweakbindingaffinityVHLE3ligaseligands[J].JMedChem,2019,62(24):11218-11231.
[57]LeeGT,NagayaN,DesantisJ,etal.EffectsofMTX-23,anovelPROTACofandrogenreceptorsplicevariant-7andandrogenreceptor,onCRPCresistanttosecond-lineantiandrogentherapy[J].MolCancerTher,2021,20(3):490-499.
[58]ChenL,HanL,MaoS,etal.DiscoveryofA031aseffectiveproteolysistargetingchimera(PROTAC)androgenreceptor(AR)degraderforthetreatmentofprostatecancer[J/OL].EurJMedChem,2021,216:113307[2023-06-12].https://doi.org/10.1016/j.ejmech.2021.113307.
[59]TakwaleAD,JoSH,JeonYU,etal.Designandcharacterizationofcereblon-mediatedandrogenreceptorproteolysis-targetingchimeras[J/OL].EurJMedChem,2020,208:112769[2023-06-12].https://doi.org/10.1016/10.1016/j.ejmech.2020.112769.
[60]NeklesaT,SnyderLB,WillardRR,etal.ARV-110:anoralandrogenreceptorPROTACdegraderforprostatecancer[J/OL].JClinOncol,2019[2023-06-12].https://ascopubs.org/doi/10.1200/JCO.2019.37.7_suppl.259.
[61]PetrylakDP,GaoX,VogelzangNJ,etal.First-in-humanphaseIstudyofARV-110,anandrogenreceptor(AR)PROTACdegraderinpatients(pts)withmetastaticcastrate-resistantprostatecancer(mCRPC)followingenzalutamide(ENZ)and/orabiraterone(ABI)[J/OL].JClinOncol,2020[2023-06-12].https://ascopubs.org/doi/abs/10.1200/JCO.2020.38.15_suppl.3500.
[62]GaoX,BurrisIIIHA,VukyJ,etal.Phase1/2studyofARV-110,anandrogenreceptor(AR)PROTACdegrader,inmetastaticcastrationresistantprostatecancer(mCRPC)[J/OL].JClinOncol,2022[2023-06-12].https://ascopubs.org/doi/abs/10.1200/JCO.2022.40.6_suppl.017?af=R.
[63]PetrylakDP,StewartTF,GaoX,etal.Aphase2expansionstudyofARV-766,aPROTACandrogenreceptor(AR)degrader,inmetastaticcastration-resistantprostatecancer(mCRPC)[J/OL].JClinOncol,2023[2023-06-12].https://ascopubs.org/doi/abs/10.1200/JCO.2023.41.6_suppl.TPS290.
[64]NIH.AndrogenreceptordegraderCC-94676,2022[EB/OL].(2023-03-06)[2023-06-12]https://www.cancer.gov/publications/dictionaries/cancer-drug/def/androgen-receptor-degrader-cc94676.
[65]KimGY,SongCW,YangYS,etal.Chemicaldegradationofandrogenreceptor(AR)usingbicalutamideanalog-thalidomidePROTACs[J/OL].Molecules,2021,26(9):2525[2023-06-12].https://doi.org/10.3390/molecules26092525.
[66]LiangJJ,XieH,YangRH,etal.Designed,synthesizedandbiologicalevaluationofproteolysistargetingchimeras(PROTACs)asARdegradersforprostatecancertreatment[J/OL].BioorgMedChem,2021,45:116331[2023-06-12].https://doi.org/10.1016/j.bmc.2021.116331.
[67]HanX,ZhaoL,XiangW,etal.Strategiestowarddiscoveryofpotentandorallybioavailableproteolysistargetingchimeradegradersofandrogenreceptorforthetreatmentofprostatecancer[J].JMedChem,2021,64(17):12831-12854.
[68]XiangW,ZhaoL,HanX,etal.DiscoveryofARD-2585asanexceptionallypotentandorallyactivePROTACdegraderofandrogenreceptorforthetreatmentofadvancedprostatecancer[J].JMedChem,2021,64(18):13487-13509.
[69]KadmielM,CidlowskiJA.Glucocorticoidreceptorsignalinginhealthanddisease[J].TrendsPharmacolSci,2013,34(9):518-530.
[70]PalitSAL,VisD,StellooS,etal.TLE3lossconfersARinhibitorresistancebyfacilitatingGR-mediatedhumanprostatecancercellgrowth[J/OL].Elife,2019,8:e47430[2023-06-12].https://doi.org/10.1038/s41598-022-05753-3.
[71]NarayananS,SrinivasS,FeldmanD.Androgen-glucocorticoidinteractionsintheeraofnovelprostatecancertherapy[J].NatRevUrol,2016,13(1):47-60.
[72]SerritellaAV,ShevrinD,HeathEI,etal.PhaseI/IItrialofenzalutamideandmifepristone,aglucocorticoidreceptorantagonist,formetastaticcastration-resistantprostatecancer[J].ClinCancerRes,2022,28(8):1549-1559.
[73]RosetteC,AganFJ,RosetteN,etal.ThedualandrogenreceptorandglucocorticoidreceptorantagonistCB-03-10aspotentialtreatmentfortumorsthathaveacquiredGR-mediatedresistancetoARblockade[J].MolCancerTher,2020,19(11):2256-2266.
[74]WuM,XieY,CuiX,etal.Rationaldrugdesignforandrogenreceptorandglucocorticoidsreceptordualantagonist[J/OL].EurJMedChem,2019,166:232-242[2023-06-12].https://pubmed.ncbi.nlm.nih.gov/30711833/.
[75]ChaiX,HuXP,WangXY,etal.Computationallyguideddiscoveryofnovelnon-steroidalAR-GRdualantagonistsdemonstratingpotencyagainstantiandrogenresistance[J/OL].ActaPharmacolSin,2023:1-19[2023-06-12].https://pubmed.ncbi.nlm.nih.gov/36639570/.DOI:10.1038/s41401-022-01038-7.
美编排版:陈鑫茹
感谢您阅读《药学进展》微信平台原创好文,也欢迎各位读者转载、引用。本文选自《药学进展》2023年第8期。