
《储能科学与技术》推荐|锂金属负极固态电解质界面膜形成和生长机理的理论研究进展
作者:周国兵1,2(),许审镇1()
单位:1.北京大学材料科学与工程学院;2.江西师范大学化学工程学院,江西南昌330022
引用:周国兵,许审镇.锂金属负极固态电解质界面膜形成和生长机理的理论研究进展[J].储能科学与技术,2024,13(9):3150-3160.
DOI:10.19799/j.cnki.2095-4239.2024.0586
本文亮点:1.本文综述了不同理论模拟方法在SEI结构、组分和生长过程的最新研究进展;2.本人提出可结合机器学习和动力学蒙特卡罗方法来实现长时域SEI形成和生长过程模拟的技术方案。
摘要锂金属负极因其极高的理论比容量在锂离子电池领域引起了极大的关注,但其高反应活性会引发电解液组分发生一系列复杂的降解反应,并在电极表面生成固态电解质界面膜(SEI)。SEI钝化层一方面能抑制电解液持续损耗,另一方面也会显著影响电池的循环性能。因此,从原子/分子层面阐明SEI形成和生长机理成为了近些年的研究重点和热点。本文综述了不同理论模拟方法在SEI结构、组分和生长过程的最新研究进展,介绍了经典分子动力学、反应力场分子动力学、第一性原理分子动力学、机器学习力场分子动力学以及动力学蒙特卡罗等模拟方法在SEI研究中的成功案例。讨论了现有理论计算方法在模拟SEI形成和生长机理方面的局限性,提出可结合机器学习和动力学蒙特卡罗方法来实现长时域SEI形成和生长过程模拟的技术方案展望。
关键词固态电解质界面膜;形成和生长机理;分子动力学;动力学蒙特卡罗;机器学习
锂离子电池(LIB)因其高能量密度、长循环寿命和高效率等优势在便携式电子设备、电动汽车以及大规模储能等领域得到了广泛的应用。目前,以石墨为负极的传统LIB已经接近其理论容量(372mAh/g),无法满足各种电子设备日益增长的储能需求。锂金属因其极高的理论比容量(3860mAh/g)和极低的电化学还原电位(-3.04Vvs.标准氢电极)被认为是新一代LIB的理想负极材料。然而,锂金属负极的大规模商业化仍存在诸多的问题和挑战,比如电极表面不均匀的锂沉积、循环过程中巨大的体积变化等。这些问题主要与锂金属电极/电解液界面结构和性质有关。在电池循环过程中,锂金属与电解液会发生一系列复杂的化学和电化学反应并在锂金属电极表面形成固态电解质界面膜(SEI)。SEI不仅能够有效抑制锂金属与电解液之间的寄生反应,同时还会影响锂的沉积和剥离行为,进而影响LIB的库仑效率、安全性能和循环寿命。因此,深入理解SEI的组成、结构、形成和生长机理对于实现锂金属负极的大规模应用和推动下一代高能量密度可充电电池技术的发展具有重要意义。
近年来,研究人员采用多种实验表征技术探究了SEI的结构和组成,包括X射线光电子能谱(XPS)、傅里叶变换红外光谱(FTIR)、拉曼光谱(Raman)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)以及原子力显微镜(AFM)等。这些技术在纳米尺度上揭示了SEI是一种多层的“马赛克”结构,主要由致密的无机内层(LiF、Li2O、Li2CO3、LiOH、LiH、Li2S、Li3N等)和多孔的有机外层[ROCO2Li、ROLi、(CH2OCO2Li) 2等]组成。尽管这些技术在表征SEI的结构方面取得了显著进展,但在揭示SEI形成机理方面仍存在局限性。SEI的形成是一个动态过程,涉及电解液的(电)化学分解、锂离子的迁移和电子的交换,这些过程在时间和空间上的复杂性使得实时监测SEI的形成和演变极具挑战。此外,SEI的化学组成和结构可能因电解液的类型、浓度、温度、电流密度等实验条件的不同而有所差异,这增加了对SEI形成机理进行普适性描述的难度。因此,仅仅依靠当前的实验表征技术来探究SEI的生长和形成机理显然是不够的。理论计算能够在原子水平上帮助实验科学家理解各种复杂过程的微观结构和动态信息,已被广泛用于模拟研究SEI的生长和形成过程。本文首先总结了传统理论计算方法在研究SEI方面的最新进展,最后对机器学习方法在研究SEI中的应用前景进行了展望。
1分子动力学方法在SEI中的研究进展
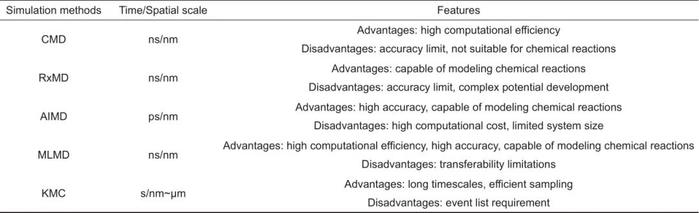
目前,研究SEI的理论计算方法主要有经典力场分子动力学(CMD)、反应力场分子动力学(RxMD)、第一性原理分子动力学(AIMD)以及机器学力场分子动力学(MLMD)。这些方法在模拟SEI的形成和生长过程中具有各自的优势和适用性(表1)。
表1 用于SEI研究的不同理论模拟方法对比
1.1 经典力场分子动力学(CMD)
CMD方法是研究物质微观结构和性质的重要工具之一。它的基本思路是将原子或分子看作相互作用的质点,通过数值积分求解牛顿运动方程获得粒子的运动轨迹。在CMD模拟过程中,粒子之间的相互作用包含成键和非键两部分,其中非键部分包含范德华和静电相互作用,通常采用Lennard-Jones和库仑势进行描述,势函数中的参数可通过拟合量子化学计算或实验数据得到。CMD模拟方法适用于研究从简单的气体分子到复杂的生物大分子等各种体系的动力学行为,目前已被广泛用于研究电极/电解液界面SEI的微观结构。Sui等采用CMD模拟方法研究了3种不同的聚(1,3-二氧戊环)(PDOL)基电解液(PDOL-in-DOL、DOL-in-PDOL和高浓度PDOL-in-DOL)在锂金属负极的界面结构,发现DOL-in-PDOL和高浓度PDOL-in-DOL电解液中的聚合物分子能聚集在锂金属表面,使得这两个体系具有更稳定的界面结构。另外,PDOL-in-DOL体系界面锂离子的溶剂化结构包含DOL分子并分布在锂金属表面,而DOL-in-PDOL体系界面锂离子的溶剂化结构主要由聚合物分子和TFSI-阴离子构成[图1(a)]。Lourenço等采用CMD模拟方法研究了3种不同的离子液体[C2mim][123Triaz]、[C2mim][TFSI]和[P222mom][TFSI]分别与聚环氧乙烷(PEO)和锂盐组成的电解液在锂金属负极的界面结构。模拟结果表明3种电解液在锂金属表面均存在明显的离子聚集现象,而界面结构和组成则与离子液体的组分相关。在离子液体的离子尺寸较小的电解液体系中,锂金属表面会有更高的PEO密度分布,锂离子也有更高的扩散系数;当离子液体的离子尺寸较大时(比如[P222mom]+阳离子),PEO和锂离子在锂金属表面的密度分布较低[图1(b)]。前期这些研究结果说明锂金属负极的界面结构与电极液组分相关,选择合适的电解液组分可以调节锂金属负极/电解液界面结构和组成,控制电极表面的SEI形成,提高电池性能。值得注意的是,CMD方法虽然能够模拟SEI的微观结构,但是其原子间相互作用的描述方式是基于参数优化的经验力场构建,并不是基于第一性原理计算获得,使得其无法描述SEI形成过程中复杂的化学反应。
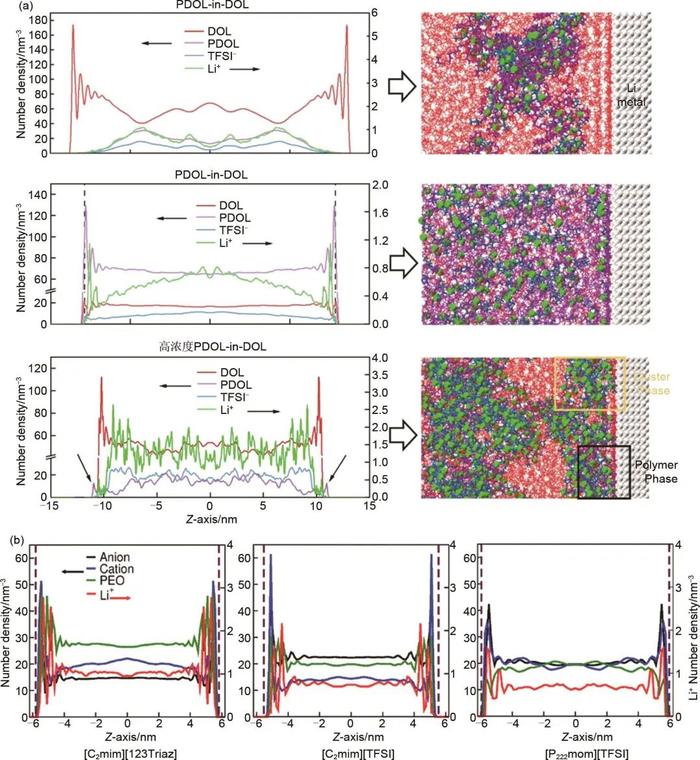
图1(a)锂金属表面PDOL-in-DOL,DOL-in-PDOL和高浓度PDOL-in-DOL三种不同体系中电解液组分的密度分布和结构;(b)[C2mim][123Triaz]、[C2mim][TFSI]和[P222mom][TFSI]体系中不同电解液组分的密度分布
1.2 反应力场分子动力学(RxMD)
RxMD是一种基于反应力场(ReaxFF)的分子动力学模拟方法。与传统力场不同,ReaxFF使用了一套基于键序的力场,这使得它能够动态模拟化学键的断裂和形成过程。RxMD的核心思想是使用一组参数化的解析函数来描述原子间的各种相互作用,包括共价键、离子键、范德华力、静电力等。这些函数依赖于原子间的距离、键级、原子类型等因素。通过合理设定这些参数,ReaxFF能够捕捉化学反应过程中动态演化的电荷变化,从而可以模拟复杂的化学反应过程。ReaxFF参数可以根据不同的化学体系优化,目前已被广泛用于研究锂金属负极表面SEI的生长和形成过程。Pao等利用RxMD与EChemDID结合的方法研究了充放电循环过程中不同电解液组成对SEI膜形成和演变的影响,发现无论电解液组成如何,在循环充电周期中都会出现不均匀的锂还原现象[图2(a)]。含氟电解液添加剂可以通过形成密集的SEI或抑制电解液分解显著减轻锂金属负极的粗糙化过程。Cheng等采用RxMD和AIMD结合的方法研究了不同浓度的有机电解液(DOL+LiFSI)在锂金属负极表面的还原分解反应及SEI形成过程。研究结果表明,随着盐浓度的增加,SEI的组成从溶剂衍生层转变为盐衍生层。在低浓度条件下,FSI-阴离子和DOL溶剂的完全分解有助于SEI膜无机和有机组分的形成,而在高浓度条件下,FSI-阴离子发生不完全分解反应,通过初始S-F键断裂消耗自由Li0,这将抑制DOL溶剂发生牺牲性还原反应。此外,他们还研究了DOL与LiPF6组成的有机电极液在锂金属负极表面形成SEI的反应机理,发现PF6-阴离子在接触锂金属负极时可以通过还原反应完全分解[图2(b)],或者在电解液体相中转化为PF5,其中分解产物(F-和Px-)将形成SEI膜的无机部分,而PF5可以作为DOL聚合的引发剂。尽管RxMD方法能够描述电解液分子与锂金属负极之间的反应过程,但其仍存在一些局限性。比如:ReaxFF的参数化过程非常复杂且耗时。另外,RxMD相比CMD的计算成本更高,使得RxMD方法只能达到纳秒量级的模拟时间尺度,无法模拟完整的SEI形成过程。
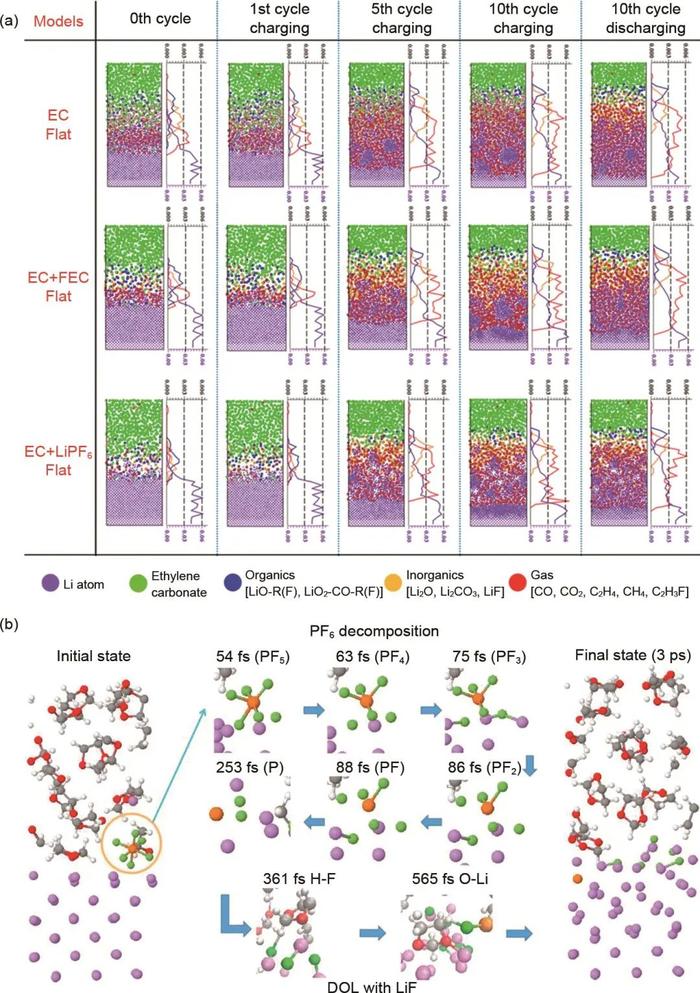
图2(a)充放电循环过程中不同锂金属电极/电解液界面SEI中化学物质的分布;(b)锂金属表面PF6-阴离子和DOL分解的反应快照图
1.3 第一性原理分子动力学(AIMD)
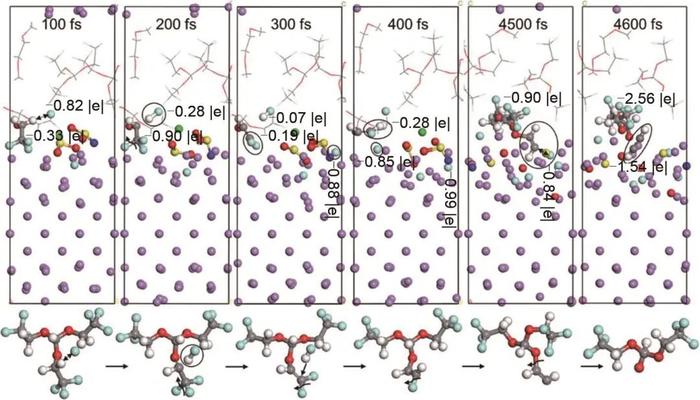
AIMD是一种结合量子力学和经典分子动力学的模拟方法,能够在原子层面上揭示物质的结构、性质和反应动力学。与基于经验势函数的CMD和RxMD不同,AIMD通过第一性原理计算原子间的相互作用,因此可以准确模拟电解液在锂金属负极表面的分解和SEI的形成过程。Merinov等采用AIMD模拟方法研究了[Pyr14][TFSI]离子液体在锂金属负极表面形成SEI膜的机理,模拟结果表明Pyr14+阳离子保持稳定并远离锂金属表面,而TFSI-阴离子与锂金属相互作用并迅速分解。同样,Kashyap等采用AIMD模拟方法研究了在超浓电解液LiTFSI/乙腈(AN)和锂金属负极界面处SEI组分的形成机制及其分布,发现初始SEI膜的形成主要来源于TFSI-阴离子的还原分解,而AN分子在锂金属附近的氧化还原过程中保持稳定。此外,Seminario等采用AIMD模拟方法研究了基于三甲基磷酸酯(TMP)和LiFSI电解液在锂金属负极表面生成SEI的形成和演变,研究发现第一个FSI-阴离子在与锂金属负极接触时完全解离,形成了Li2O、LiF、Li2S和Li3N等锂的二元组分。随后在第二次FSI-阴离子解离期间形成了Li2S、Li2O、LiF、Li3NSO2,在第三次解离期间形成了Li2SO2NSO2和LiF。SEI的形成将减弱随后靠近表面的FSI-阴离子的分解,这清楚地表明了SEI对锂金属负极的保护机制。Balbuena等结合CMD和AIMD方法研究了锂金属负极表面LiFSI锂盐/二甲氧基乙烷(DME)/三(2,2,2-三氟乙基)正甲酸酯(TFEO)混合电解液的反应活性,结果表明当LiFSI和TFEO彼此靠近且接近锂金属表面时,FSI-阴离子的分解可以引发一系列反应,导致TFEO分解,并在锂金属表面形成SEI(图3)。然而,如果FSI-阴离子和DME分子与锂离子形成复合物,则FSI-阴离子的稳定性显著增加。需要指出的是,虽然AIMD方法能够准确描述电解/电解液界面处的复杂化学反应,但其计算成本非常高,每个动力学步骤都需要进行复杂的量子力学计算,这极大限制了可模拟的体系大小和时间尺度。通常,AIMD模拟仅限于几百个原子和几十皮秒的时间尺度,使得该方法无法捕捉SEI形成的全过程。
1.4 机器学习力场分子动力学(MLMD)
MLMD是一种基于机器学习力场(MLFF)的分子动力学模拟方法,基本思想是利用机器学习算法对大量的分子结构和相互作用数据进行训练,建立一个能够准确预测分子体系的势能函数模型,从而替代传统的经验力场,并用于模拟分子体系的结构和动力学行为。相较于传统的经验力场,MLFF能够提供更准确的相互作用描述,尤其是对于化学反应、催化过程、溶剂效应等需要高精度描述的情况。目前,MLMD在化学、材料、能源、物理等领域展现出广泛的应用前景。Yang等结合密度泛函理论(DFT)计算和机器学习方法开发了一套深度势能模型用于揭示锂金属负极/固态电解质β-Li3PS4界面SEI形成和演化的复杂机制。模拟结果表明Li|β-Li3PS4界面存在四阶段的演化过程:①快速离子扩散,②成核,③Li2S生长和④稳定化[图4(a)~(e)]。进一步分析结果表明Li|β-Li3PS4界面形成的SEI可以分为晶体和非晶区域[图4(f)],300K条件下形成SEI的厚度约为12.4nm,当温度升高至600K时SEI厚度达到约20.6nm。Wang等提出了一种稳定性指示采样(SIS)的算法用于开发锂金属负极/电解液(LiFSI+DOL)界面体系的MLFF,通过结合开发的MLFF和分子动力学模拟方法再现了锂金属电池中一些已知的SEI成分,包括LiF、Li2O、LiOH等。此外,研究还发现在1mol/L低浓度界面体系中存在LiF的离子聚集结构,而在10mol/L高浓度界面体系中则观察到完全的S-F断裂以及不完全的N-S断裂,但未观察到溶剂分子DOL发生分解。虽然MLMD方法同时具备了CMD方法的计算效率和AIMD方法的计算精度,但是其模拟时间尺度同样局限在纳秒级别,无法达到SEI生长所需的时间尺度。此外,SEI生长过程涉及复杂的化学反应网络,MLMD在捕捉这些复杂反应路径和中间态方面仍然存在困难。尽管机器学习模型可以拟合势能面,但对反应机制的精确描述和反应路径的预测仍然具有挑战性,尤其是在反应网络高度复杂的情况下。
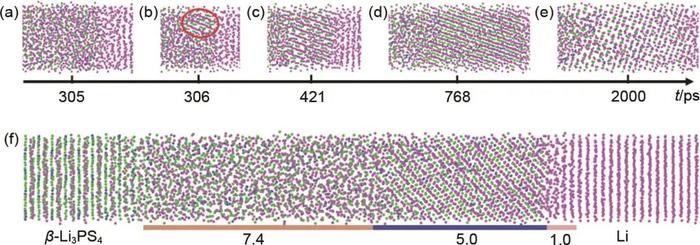
图4Li|β-Li3PS4界面模型的 (a) 离子扩散;(b) 和 (c) 成核;(c) 和 (d) 晶体生长;(d) 和 (e) 稳定结构;(f)Li|β-Li3PS4界面模型的最终结构
2动力学蒙特卡罗(KMC)在SEI膜中的研究进展
KMC是基于马尔科夫过程的随机模拟方法,可用于模拟各类复杂的动态过程,通过模拟粒子在时间和空间上的随机运动,来探究体系的演化行为和动力学特性。在每个KMC步骤中,根据事件发生概率随机选择一个事件发生,并相应更新系统的状态。通过重复这一过程,KMC模拟可以有效跟踪复杂体系在长时间尺度上的演化。不同于传统的CMD模拟,KMC模拟只关注系统中各种可能发生的事件(如化学反应、相变、扩散等),不需要追踪原子/分子的运动轨迹,这使得KMC模拟具有更高的计算效率,并且可以模拟跨时间尺度从纳秒到数小时的过程。SEI的形成机理涉及多种复杂的化学反应和物理过程,如电解液分解、离子迁移、相转变等。这些过程的时空尺度差异较大,给传统动力学模拟方法带来了很大挑战。KMC模拟在时间和空间尺度上具有很强的扩展能力,可以用于研究从纳米级别的初始反应到微米级别的膜厚度变化,以及从初始成膜到长期循环稳定性的演化过程,因此KMC在研究SEI的宏观行为和微观机制方面具有独特优势。
2.1 二维晶格模型
Wenzel等构建了一套包含化学反应、扩散和聚集过程的反应网络,并结合KMC方法从纳米尺度模拟SEI的时空演变。模拟结果表明SEI的形成和生长通过溶液介导途径进行,SEI前体在远离表面的溶液中通过成核过程聚集,随后迅速生长形成多孔层,最终覆盖表面。另外,他们还发现SEI的厚度与发生成核反应位置有关,当成核反应远离表面发生时,生长的SEI最稳定。Kwon等采用KMC方法研究了SEI层中锂离子的扩散能垒对锂枝晶生长的影响,发现锂离子的扩散能垒不同,SEI的表面粗糙度也不同。锂离子的扩散能垒越低,其扩散速率越高,导致在锂金属负极-SEI界面附近有高的锂离子通量,发生快速且不均匀的锂还原[图5(a)、(b)]。此外,他们还探究了两种不同机械强度的SEI组分(LiF和ROLi)对锂枝晶生长的影响,发现当SEI为LiF时,锂能均匀沉积在锂金属表面,而当SEI为ROLi时,锂金属表面会形成锂枝晶[图5(c)、(d)]。这主要是因为ROLi的杨氏模量(YROLi≪6GPa)很小,使得ROLi难以承受枝晶诱导的应力。因此,沉积的锂金属可以很容易地穿透ROLi并不规则地生长。LiF具有较大的杨氏模量(YLiF≫6GPa),这使其能够在破裂或应变之前承受大量的应力。在这种情况下,新沉积的锂金属很难穿透LiF。
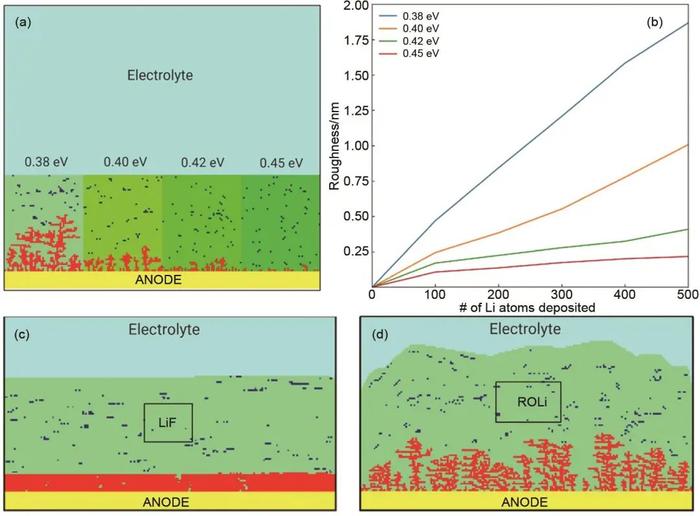
图5(a)具有不同锂扩散能垒的SEI各区域中锂枝晶形成的快照图;(b)SEI不同区域的粗糙度演化趋势图;(c)LiF中锂均匀沉积的快照图;(d)ROLi中锂枝晶生长的快照
2.2 三维晶格模型
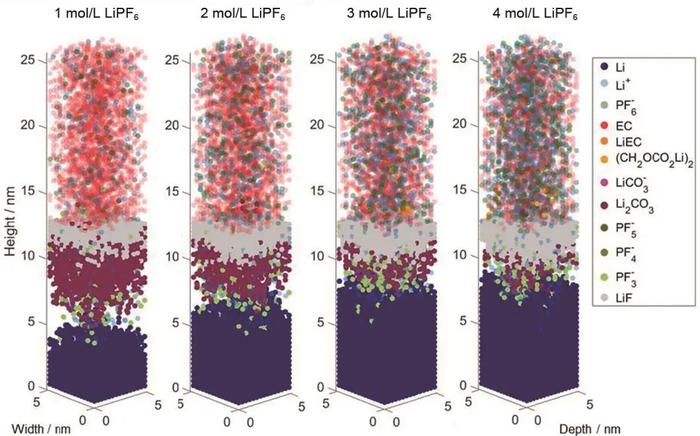
Krewer等结合分子动力学(MD)、密度泛函理论(DFT)和KMC探究了2mol/LLiPF6/碳酸乙烯酯(EC)电解液在锂金属模型表面形成SEI过程中的内部物种分布。首先,利用MD和DFT产生KMC模拟需要的参数,然后采用KMC方法模拟三维锂金属模型表面SEI的生长过程。发现表面形成的SEI具有层状结构,从表面开始依次为无机层和有机层,其中无机层包含Li4F和Li2CO3,而有机层产物为(CH2OCO2Li)2。通过分析SEI形成的锂来源,认为表面形成的SEI本质上是由于电解液引起的锂金属腐蚀过程,只有一小部分来自于电解液中的盐分解。最近,该团队通过进一步耦合KMC与连续介质模型发展了一种多尺度模拟方法深入分析LiPF6/EC电解液中最初SEI的形成过程,发现传输是导电盐LiPF6降解的限制过程,而电解质溶剂EC的分解则受到其反应动力学的限制。分析结果显示大部分无机SEI由Li2CO3和锂金属上方的LiF组成,通过改变电解质中的盐浓度,可以调节SEI无机层中Li2CO3和LiF的比例以及无机层的厚度(图6)。此外,他们还发现锂离子浓度和溶剂环境对形成的SEI形态有巨大影响,高锂离子浓度导致形成层状结构的无机SEI;而低局部锂离子浓度则形成更类似马赛克结构的无机SEI和更多的有机SEI物种。
3结语与展望
SEI是电解液在LIBs负极表面发生一系列化学和电化学反应而形成的钝化层,SEI的结构和性质对电池的性能起着至关重要的作用,尤其是在锂金属被用作高能量密度的电池系统中。SEI的形成涉及非常复杂的物理和化学变化过程,这些过程在时间和空间上的复杂性,使得对SEI形成机理的全面理解极具挑战性。近年来,理论计算在揭示SEI的微观结构和动态形成过程方面取得了显著进展,为解决锂金属负极在高能量密度电池中的应用难题提供了理论基础。理论模拟结果不仅能够提供实验难以直接观测的微观细节,同时可以预测不同电解液和添加剂对SEI形成的影响,指导实验选择和优化电解液配方,减少实验筛选的范围和成本。然而,需要指出的是,SEI的形成过程涉及从原子级到微米级的多个空间尺度,而且时间尺度也达到秒级以上,常见的理论模拟方法(如CMD、RxMD、AIMD和MLMD)无法达到SEI生长所需的空间和时间尺度。KMC模拟方法克服了上述模拟方法的局限性,同时保留了详细的分子信息,可以有效处理复杂的反应网络和长时间的演化过程,已经广泛用于模拟SEI的生长过程。当前研究通常采用传统KMC模拟SEI生长,这种方法需要通过实验或理论计算预先确定所有可能的反应路径和对应的速率常数,并将其存储在一个数据库中。基于预定义的反应路径和速率数据库,生成当前状态下可能的反应事件列表。根据反应速率常数,通过蒙特卡罗算法选择下一个要发生的事件,根据选定事件的速率,推进体系的模拟时间。然而,电池循环过程中电极/电解液界面处的局域环境变化很大,不同的局域环境会影响反应事件的能垒,因此基于提前构建反应事件列表的传统KMC方法难以准确模拟SEI的真实生长过程。相较于传统KMC,on-the-flyKMC(OTF-KMC)是一种更为先进的计算方法,它在模拟过程中能够实时构建反应事件列表。具体而言,OTF-KMC在模拟过程中能够动态生成反应路径和速率常数。这意味着每当体系状态发生变化时,新的反应路径和速率常数会实时计算并更新。这种方法避免了预先定义的数据库可能带来的不准确性和不完整性。此外,通过实时计算和更新反应路径,OTF-KMC能够灵活适应体系的动态变化,捕捉新的反应途径和速率,提供更高的模拟精度。因此这种方法特别适用于研究复杂的、具有未知反应路径的体系,比如SEI的生长过程模拟。然而,OTF-KMC方法需要精确的原子间作用势才能获得可靠的模拟结果,传统的经验力场难以满足这一要求。MLFF是一种基于机器学习算法训练第一性原理计算数据的新兴分子力场模型,该力场能够克服传统经验力场精度不足和从头计算方法计算成本高的问题,在保持高精度的同时大幅提高计算效率。因此,未来的研究可以将MLFF与OTF-KMC整合用于模拟SEI的生长过程。这种新的研究范式不仅能够保持高精度,还能显著扩展模拟的时间和空间尺度,有助于研究人员更全面地捕捉SEI形成和演化的各个层面,为深入理解和优化SEI提供更强大的工具。另外,通过MLFF/OTF-KMC模拟还可以产生大量不同局域环境下的反应事件的能垒,将这些反应事件作为数据集,同时结合机器学习算法可以训练得到一个机器学习模型。最后,可以使用生成的机器学习模型预测锂金属电极/电解液界面处不同区域反应事件的能垒,从而实现空间尺度在微米级的SEI的生长过程模拟。总之,机器学习与多尺度模拟方法的结合将为我们提供新的可能,有望在深入理解SEI的形成机制以及指导SEI层优化设计等方面取得重大突破。
第一作者:周国兵(1989—),男,博士,副研究员,主要研究方向为材料模拟与理论催化,E-mail:gbzhou@jxnu.edu.cn。
通讯作者:许审镇,博士,助理教授,主要研究方向为电化学理论计算与方法开发,E-mail:xushenzhen@pku.edu.cn。

邮发代号:80-732
联系热线:010-64519601/9602/9643
投稿网址:http://esst.cip.com.cn/CN/2095-4239/home.shtml