
FDA伴随诊断监管政策的演变(下)
转自:药渡
近日,泛生子首席医疗官胡云富博士在DIA2023年会伴随诊断和精准医疗专场分享了题为《FDA伴随诊断监管政策的演变与案例研究》(TracingtheEvolutionofFDACDxApprovalPoliciesThroughCaseStudies)的报告。胡云富博士为前美国FDA资深注册评审专家,在FDA任职11年期间,曾带领团队完成诸多里程碑式的伴随诊断(CDx)产品和服务审批工作。
本次报告从FDA监管CDx的基本原则、CDx监管政策的演变历程、美国LDT和CDx监管的现状与未来三个方面进行了分享。本篇将介绍报告的第三部分。


泛生子首席医疗官胡云富博士
第三篇章:美国的LDT和CDx监管
美国现行LDT监管体系
LDTs(LaboratoryDeveloped Tests)一般是指在实验室内部研发、验证和使用的检测方法。在美国,医疗保险和辅助服务中心(CMS)依据临床实验室改进修正案(CLIA‘88)对临床实验室进行监管,根据技术复杂度分类进行CLIA认证。CMS对临床实验室的人员专业性、质控能力、产品验证及实验记录等方面制定了相关标准和要求。
临床实验室使用的IVD试剂盒是经FDA审批的;但美国实验室自主研发试剂(LDT)大多未经FDA审批,不对外进行商品化的售卖,限于在实验室内部由专业人员开展使用。
美国LDT监管改革的尝试
尽管CMS要求NGS检测需要作为CDx获批才能给予医保报销,但实际临床实验室使用的检测以未经FDA审批的LDTs居多,尤其对于临床具有高风险的检测,其结果的严谨性和准确性将影响临床决策判断。FDA很早就注意到此现象,在2010年宣布有意重新考虑对LDTs的自由裁量政策,并在2014年发布了LDT监管指南的初步草案,基于风险等级提出不同监管措施,重点加强对高风险LDTs的监管。
该草案的发布引发了业界广泛的讨论,FDA综合收集反馈意见,于2017年发布了关于LDT的讨论文章,提出重点关注新的和有显著调整的高、中风险LDT的上市前审批、质量体系和注册备案要求;在CLIA质量体系基础上增加设计开发控制(Designcontrol)、纠正预防措施(CAPA,CorrectiveActionandPreventiveAction)等要求。
相比于指南和审批案例,立法途径具有更高强制执行力。2020年、2021年,VALID法案(VerifyingAccurateLeading-edgeIVCTDevelopmentAct)两次被提案至国会,法案提出授予FDA审查、批准IVCT(体外临床检测,invitroclinicaltests)的权力,遵从基于风险的监管框架,其中IVCT包括IVD试剂盒与LDT。延续2014年FDA指南草案的要求,VALID法案提出所有IVD制造商和临床实验室都需要遵守“IVD”要求,包括在FDA注册备案、质量要求、不良事件报告等;高风险检测需要进行上市前审批,低风险检测可以通过技术认证或“pre-cert”途径上市。
为保障患者利益而持续努力
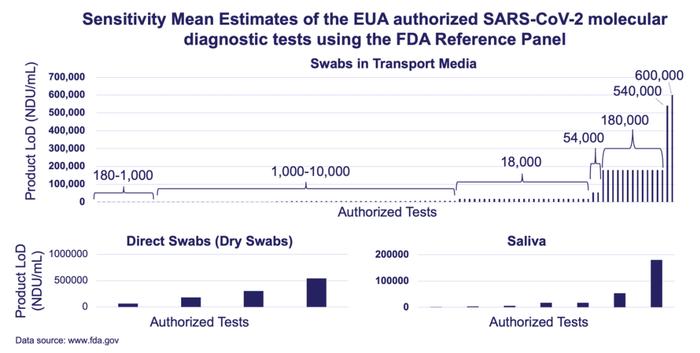
临床决策依赖于临床检测的准确性。在美国当前标准下,LDT的准确性没有得到充分监督,可能会危及患者安全。这一风险在疫情期间通过EUA(EmergencyUseAuthorization,紧急使用授权)的COVID-19检测性能上得以侧面反映。在一项FDA发起的盲测中,不同COVID-19核酸检测的最低检出限(LoD)竟然表现出高达上千倍的差异,据估算由此会导致一部分阳性样本漏报[1][2]。
不同COVID-19核酸检测的最低检出限
由此可见,对临床检测,特别是高风险的检测,更严格的监管尤为必要,一个更强大的监管框架将有助于确保临床检测的安全有效进行。FDA设备和放射健康中心(CDRH)的ElizabethHillebrenner于今年年初表示,FDA会继续通过规章制定的方式推进针对LDT的合理监管。
今年6月,FDA发布了终版指南《与特定体外诊断试验共同使用的肿瘤药物:试点计划》[3],并宣布了一个新的自愿试点计划,旨在通过透明的性能建议,帮助选择用于特定肿瘤药物治疗的诊断测试。一些未经FDA审批的LDTs可能存在因性能不佳而对治疗决策产生负面影响的风险。FDA官方网站表示,该试点计划是缓解在癌症药物治疗决策中使用LDTs风险的措施之一,同时FDA将继续推进更广泛的LDTs监管方式,包括规章制定[4]。目前我们尚不清楚各项监管措施将如何协同工作,但我们期待未来在各方的不懈努力下,无论是否用于伴随诊断目的,LDTs都能为患者提供更为准确可靠的结果,特别是有未被满足的临床需求的患者。
再将视野迁回国内,不难发现,无论是2013年原国家卫生计生委医政管理局批准成立的“国家卫生计生委个体化医学检测试点单位”(也称为“LDT试点单位”)之初衷,还是2023年年初国家药监局综合司和国家卫健委联合发布的《关于开展医疗机构自行研制使用体外诊断试剂试点工作的通知》及各地方陆续发布的LDT试点文件,或是近些年NMPA对体验诊断试剂、原研伴随诊断等相关指导原则和意见征询稿的陆续发布,都在不断推动着诊断试剂产品开发和应用价值往更高水平的方向发展,经严谨性能验证且可提供精准检测结果的产品才能在临床决策指导上提供更具价值的参考作用,才能更久经考验的得到广泛认可和应用。