
从指导原则和国内外案例看小分子创新药的药学研究策略
转自:药渡
9月7日,CDE发布了《中国新药注册临床试验进展年度报告》对2022年的创新药研发进行了总结,在去年上市的新药中,上市用时在5年内的品种接近50%。从2015年医改以来,创新药开发的质量和效率一直是国内的行业焦点,由于研究的渐进性、阶段性和不确定性,在不同阶段的药学研究深度、各个阶段之间的变更桥接,都将直接影响产品开发的质量和速度。目前药审中心(CDE)对创新药已颁布超过390条指导原则,基本完善的覆盖了其开发全流程,其中和药学研究相关的指导原则超过120条。
本文梳理了指导原则对小分子创新药不同开发阶段的药学研究要求,并以国内外部分产品在临床期间的研究变更历程为案例,剖析在上市过程中的药学内容和变更桥接要求,力求在法规的指导下窥见小分子创新药开发中的药学策略制定和各阶段研究深度把控。
1
新药开发研发流程和CDE指导原则
创新药从实验室发现到上市销售通常需要先后经历药物发现、临床前研究、临床研究和放大生产研究几个阶段,期间法规的指导和约束作用随着研究的推进不断被强化。当前CDE网站公布生效的指导原则共计468条,其中与创新药研究相关的指导原则超过390条,构成了新药研究的整体蓝图。研发流程各阶段中的指导原则分布情况如图1所示。
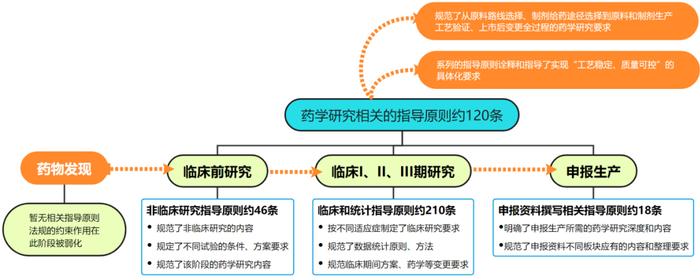
如上图所示,当前国内颁布的创新药指导原则中和药学研究相关的约120条,系统的涵盖了从原料药路线选择、晶型和盐型选择、杂质研究到制剂给药途径、剂型选择,直至生产验证、上市后变更的各个环节,力求对“工艺稳定、质量可控”进行具象化的诠释和要求。
2
药物发现阶段
药物发现阶段包括了靶点的选择、苗头化合物合成、先导化合物合成与优化,以及候选化合物(PCC)的发现。这一阶段的目的在于尽快获得活性、毒性基本满足要求的PCC,可能需要对成百上千个先导化合物进行体外活性的筛选,不同公司、不同品种具有其独特的发现过程。该阶段弱化了法规的约束作用,以确保申请人具有较高的探索灵活性,CDE的指导原则中暂未见针对此阶段的指导原则。
3
临床前研究阶段
临床前研究阶段是在人体试验开始之前,对药物合成路线、盐型晶型、处方工艺等进行适度的药学研究,并在适当的动物模型上进行安全性和有效性的评价,以评估新化合物的安全性,以确保人体试验的安全为首要目标。
本阶段的药学研究目的为:提供支持开展I期临床研究所需的初步质量可控的样品。法规的指导和规范性作用在这个阶段开始介入,确保药学、药理、以及毒理的研究内容科学、全面,以最终保障后续临床试验受试者的安全。该阶段的法规如表1所示。
表1:部分非临床研究阶段的关键指导原则
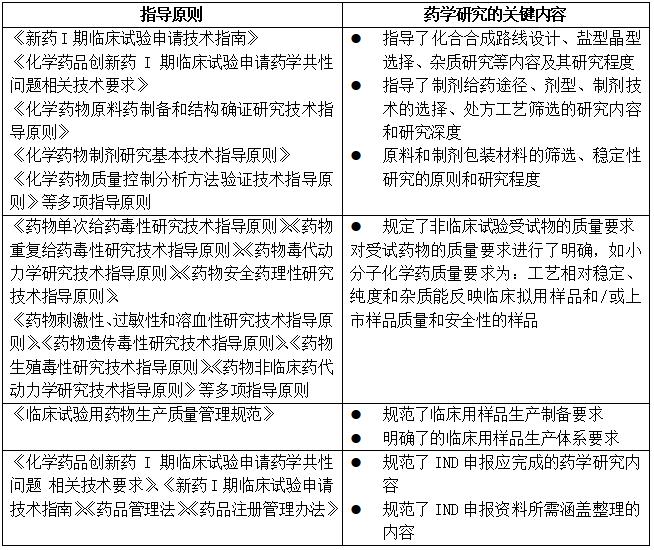
如表1所示,相较于药物发现阶段,本阶段法规和指导原则的指导和约束作用逐渐加强。针对药学研究方面,《化学药物制剂研究基本技术指导原则》、《化学药品创新药I期临床试验申请药学共性问题相关技术要求》等文件指导了该阶段原料和制剂研究的大致内容和研究程度;《药物单次给药毒性研究技术指导原则》、《药物安全药理性研究技术指导原则》等文件规定了动物评价用样品需要具备的基本药学质量:工艺相对稳定、纯度和杂质能反映临床样品/上市样品。对上述指导原则中对小分子创新药的药学研究内容和程度要求进行了梳理汇总,如图2所示。
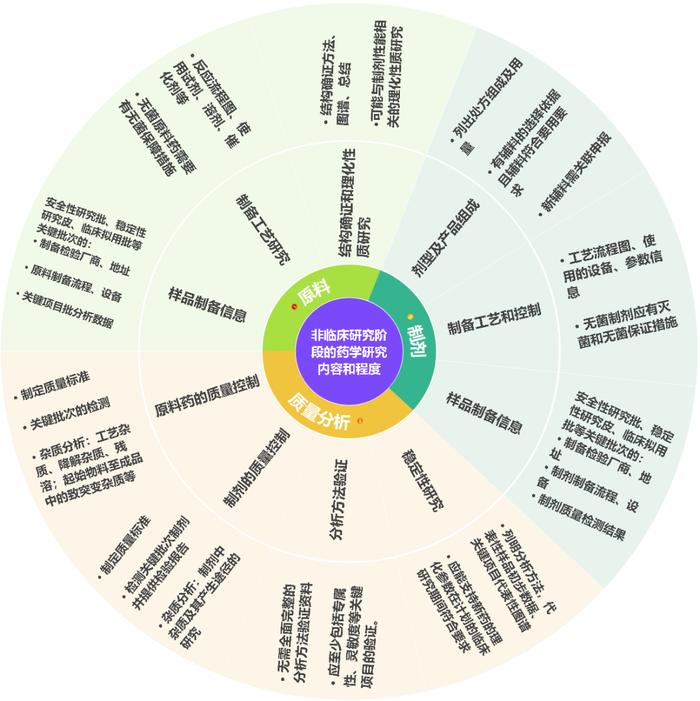
如上图所示,指导原则已然规定了此阶段基本的药学研究内容,而在此阶段药学研究的深度则需要“具体问题具体分析”。从当前国内创新研发历程看,该阶段的研发深度和PCC的理化性质、目标给药途径、竞争情况、公司资源和策略等方面息息相关,有的品种以“一次做对”为目标,比如国内已上市的泽布替尼片、伊鲁阿克片、帕米帕利胶囊等产品从临床I期到获批生产,其剂型、规格均未发生改变,也未见处方工艺变更后的体内桥接试验登记记录[1][2]。也有产品以“逐渐深入”为策略,比如国内已上市的赛沃替尼首先以一种处方工艺推进临床,后期进一步研究并变更了工艺[1][2],美国上市的奥贝胆酸片在临床前期以胶囊剂进行研究,关键临床开展前变更为片剂[3]。
4
临床研究
临床研究一般分为获批上市前的I~Ⅲ期临床,以及上市后进行的Ⅳ期临床试验,是产品能否快速注册申请成功的关键,在2015年药改之后,ICH指导原则的实施、优先审评审批政策的落地、抗癌药单臂试验等指导原则的试行,都着力于鼓励和加快临床开展的速度和质量。而在临床期间,药学的研究深度和广度不断推进,此时通常伴随了相应的变更研究,CDE发布的临床期间药学研究相关的重要指导原则如表2所示。
表2:临床期间创新药(化药)药学研究重要指导原则

从表2可知,临床期间与药学相关的指导原则围绕“变更”为主题,以变更发生的阶段、事项、品种特点等方面为考量依据,对其对比研究、桥接内容进行了规定。
以美国获批的卢曲波帕片研发历程为例,在FDA网站公布的审评材料可见[3],其I期使用的为4mg片剂,Ⅱ期使用了1mg片剂,由于两个规格的处方不同,进行了生物利用度/生物等效性研究桥接;在Ⅲ期则使用了3mg规格,由于其和I期所用片剂的处方相似,且本品为快速溶出,通过两个规格的处方和溶出比对豁免了体内桥接研究;后期增加代工厂变更生产场地时,由于拟上市制剂和Ⅲ期临床考察使用的样品处方相同,溶出相似且为快速溶出,此次也豁免了体内桥接。
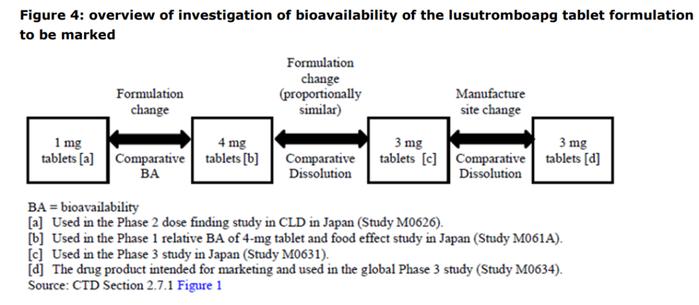
从上述变更案例可见,变更发生的时期、事项,以及产品本身的特点,共同决定了变更所需要的研究程度。在国内获批的新药产品的研究历程中也体现了临床期间的变更思路,如表3所示。
表3:国内部分创新药(化药)研发期间的变更情形[1][2]
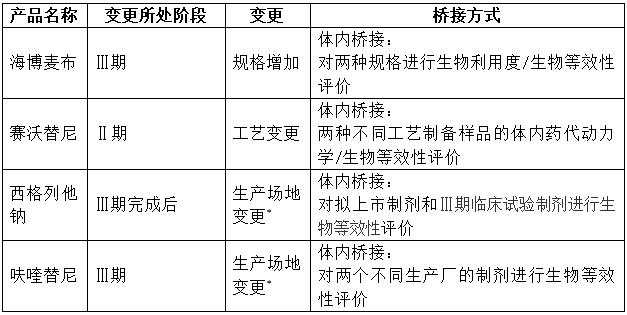
表3列举了国内部分品种在临床期间发生变更时需进行体内桥接的情形,此外,在国内艾瑞昔布、氟唑帕利、普拉替尼等几乎所有创新药的注册历程中均可见在临床开展早期的多次“补充申请”记录并无相应的体内桥接试验登记,即:如指导原则要求,越在临床后期进行的变更,需要进行体内桥接的风险也越高。
5
申报生产
在完成了药学和临床研究后,基于研究资料和数据确认产品安全、有效且质量可控后,此时向CDE提交新药上市申请。此阶段需参照ICH和国内技术指导原则对历史批次生产数据和批分析数据进行系统分析,建立疗效、安全性与产品质量的关联,完整详实的进行药学研究。相关重要指导原则如表4所示。
表4:生产研究阶段创新药(化药)药学研究重要指导原则

如表4所示,此阶段指导原则以“质量可控、工艺稳定”为目标,对如有关物质、粒度、晶型、溶出行为等与产品安全、疗效相关的关键质量属性研究要求进行了细化。以确保研究详细、深入、全面,确定稳定、重现、可商业化生产的工艺,构建完善的药品质量控制体系。
综上所述,当前药审中心(CDE)颁布的指导原则和所施行的ICH文件已经基本完善的覆盖了创新药开发的全流程,由于开发不同阶段有不同的药学研究目的,其研究策略的制定和深度的把控将对产品推进的质量和效率产生显著的影响。药学相关的指导原则也正是以不同阶段的研究内容、深度和桥接为主题的,而在实践中不同产品的药学策略又和其理化性质、制剂特性、竞争情况、公司资源等方面息息相关。
参考文献:
1、https://www.chictr.org.cn/
2、www.chinadrugtrials.org.cn
3、https://www.fda.gov/drugs