
【原创】浅谈新药Pre-IND会议资料准备
转自:注册圈
前言
2020年3月30日,国家市场监督管理总局发布的《药品注册管理办法》规定:申请人在药物临床试验申请前、药物临床试验过程中以及药品上市许可申请前等关键阶段,可以就重大问题与药品审评中心等专业技术机构进行沟通交流。
2020年12月11日,CDE发布了《药物研发与技术审评沟通交流管理办法》:首次新药临床试验申请前,申请人原则上应当向药审中心提出沟通交流会议申请,并在确保受试者安全的基础上,确定临床试验申请资料的完整性、实施临床试验的可行性。此办法发布了三个重要附件,分别为沟通交流会议申请表、沟通交流会议资料、沟通交流会议纪要模板。
2023年3月31日,CDE举办了“药品注册研发沟通交流主题线上宣讲会”,分享了沟通交流机制及常见问题等。
今天此篇文章将在沟通交流管理办法的基础上,结合本人近期的实际操作经验,详细介绍创新药首次临床试验申请前即Pre-IND会议资料的准备。不足之处,还请多多指教!
一
沟通交流会议资料
根据沟通交流管理办法,申请人准备的沟通交流会议资料应包括临床试验方案或草案、已有的药学和非临床研究数据及其他研究数据的完整资料。结合CDE沟通交流平台预约咨询申请表,申请人可参照下表准备资料。
▲ 表1-沟通交流会议资料列表
资料类别
资料内容
沟通交流会议资料
沟通交流会议PPT
预约咨询申请表;
附件1:沟通交流会议申请表;
附件2:沟通交流会议资料。
问题清单:按学科分类,包括但不限于药学、药理毒理和临床等方面的问题,每个问题应该包括背景解释,以及申请人对该问题的意见。一般情况下,一次会议拟讨论的问题不应超过10个。
药学研究资料
①原料药化合物信息、合成路线、杂质研究、质量标准;
②制剂处方、工艺研究及质量标准;
③批检验及稳定性研究情况;
④其他必要支持性材料。
此部分内容,若时间紧,可提供各部分研究总结概括,例如产品开发报告、生产工艺总结报告等。若时间充足,可提交质量综述资料2.3.S及2.3.P。
非临床研究资料
①药理学:体内外药效、安全药理学;
②药代动力学;
③毒理学;
④其他必要支持性材料。
此阶段若非临床研究报告未定稿,不必提交完整的2.4、2.6。提交的非临床总结,包括所做的非临床试验的数据、列表总结与结论。非临床研究报告可提供draft版本,不必是签字盖章版。应尽量提交IND申报时所包含的非临床研究报告,特别是毒理报告,分析方法验证报告Pre-IND可不提供。
临床资料
临床试验方案
临床开发计划
预约咨询申请表为在CDE沟通交流平台在线填写,其他资料在线上传(单个文件大小不超过20M)。以下将对沟通交流会议PPT、申请表及沟通交流问题进行详细介绍。
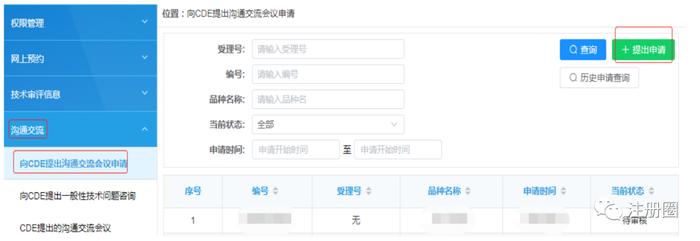
二
拟沟通问题及依据的PPT
沟通交流PPT主要内容为支持性数据总结以及讨论问题清单,另外加拟参会人员以及公司的简要介绍,创新药通常也会进行作用机制、专利、立题依据的说明。可从下表2示例的几个方面展开。
▲ 表2-沟通交流PPT主要内容
PPT项目
PPT内容
参考时间分布
1.参会人员及公司介绍
列出参会人员名单,包括职务、工作内容和工作单位,对公司进行简要介绍,让CDE了解参会人员及公司背景。
5min
2.会议目的
确保本品中已完成的临床前研究支持拟定的临床试验;确定临床试验方案及开发计划的合理性。
3.产品介绍
进行产品作用机制、专利、立题依据等的说明,让CDE了解产品的背景。
4.药学研究总结
可从以下方面介绍:
①原料药化合物信息、合成路线、杂质研究、质量标准;
②制剂处方、工艺及质量标准;
③批检验及稳定性研究情况。
10min
5.非临床研究总结
说明临床前研究(体内外药效、安全药理、药代动力学、药理毒理)的主要内容,列表说明试验结果,必要时辅以图片说明,列表可参考表3。
10min
6.临床试验方案及开发计划介绍
对研究目的、受试者人群、终点指标、对给药剂量选择、剂量组设计等进行说明。
10min
7.问题讨论
按学科顺序列出问题,如药学、非临床和临床。每个问题均应包括背景介绍以及申请人自己的意见。
25min
▲ 表3.非临床研究内容
研究类型
研究内容
试验体系/种属
给药方式/剂量
GLP依从性
试验结果
试验编号
体内药效学
XXX模型药效学研究
大鼠
口服/ xxmg/kg
否
药代动力学
PK研究
大鼠
单次口服/ xxmg/kg
多次口服 / xxmg/kg
否
28天毒理
SD大鼠28天毒性评价研究
SD大鼠
口服(QD)连续给药28天,恢复期28天
是
三
沟通交流会议申请表
沟通交流会议申请表与CDE预约咨询申请表内容大致相同,可以先填写附件1,然后在CDE平台上进行更新。其中有几个问题需要关注下:
3.1适应症:
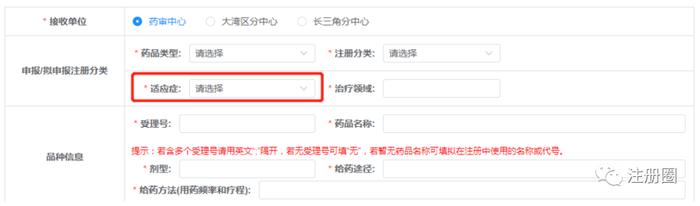
目前做自身免疫疾病领域的企业很多,有几类适应症经常会选错,例如银屑病、移植物抗宿主病(GVHD)、炎症性肠病,经常会选择为风湿性疾病及免疫药物,实际上,应分别为皮肤及五官科药物、外科及其他药物、消化系统疾病药物。适应症该如何选择呢?在药品注册申请表的填表说明中,对适应症分类进行了详细的阐述,大家可参考此处分类进行选择。

3.2拟沟通交流的学科:
新药首次申请临床试验,CDE是按照临床、药学、药理毒理3个专业来分配审核任务的,其他申请是基于申请人拟沟通交流学科来进行任务分配。
本人前期申请新药Pre-INDmeeting的时候,有一个项目只选择了临床和药理毒理专业,未勾选药学专业,CDE进行反馈的时候还是按照这三个专业来进行回复的。
另外,涉及起始剂量选择的问题,除勾选临床专业外,还涉及药理毒理专业;临床样本量设计的问题,还应勾选统计专业。下图是我们一个项目起始剂量及样本量选择的问题,CDE分三个部门:化药临床二部、药理毒理学部、统计与临床药理学部(临床药理)部进行了反馈。

3.3建议咨询时间:
一般提供3个备选时间,如果是面对面会议或者视频会议,CDE会与申请人对具体时间进行沟通。目前大部分为书面回复,这个时间也不会起到很大作用。
四
沟通交流问题
沟通交流问题要明确,需要列出具体问题,申请人提出自己的意见,这样CDE才能有针对性的进行回复。
有人会问,是否药学、药理毒理、临床3个专业都需要提出问题呢?实际上,如果这个专业研究过程中没有产生问题,也无需绞尽脑汁想出一个问题来的。当然,如上述3.2所说,新药首次申请临床试验,CDE还是会按照3个专业来进行审核反馈的。例如,我们有一个项目,药学没有提出问题,CDE首先打电话过来确认是否确实是没有问题,得到答复后对提交的资料进行了审核反馈,如下图。

以下列出了几个比较经典的问题供参考:
药学:晶型研究是否满足IND申报要求;杂质研究是否满足IND申报等;
药理毒理:非临床研究数据是否支持拟开展的临床试验;非临床研究中认定的NOAEL数据是否合理等;
临床:起始剂量的选择是否合理等。
Q&A
1.是否可以不进行沟通交流?
①申请附条件批准和/或适用优先审评审批程序的,应与药审中心沟通交流确认后,方可向国家药品监督管理局递交药品上市许可申请。
②对于技术指南明确、药物临床试验有成熟研究经验,申请人能够保障申报资料质量的,或国际同步研发的国际多中心临床试验申请,在监管体系完善的国家和/或地区已获准实施临床试验的,申请人可不经沟通交流直接提出临床试验申请。
③首次新药临床试验申请前、预防用和治疗用生物制品上市许可申请前,申请人原则上应当进行沟通交流,对于此类沟通交流情形,强调体现申请人的主体责任,在实施通知中明确告知申请人如认为无需沟通,可在申报资料中对无需沟通理由作出说明并承诺自担风险。可在申报资料1.6沟通交流会议资料中说明理由,提交承诺书。
2.Pre-INDmeeting反馈需要多长时间?
新药临床试验申请前会议为Ⅱ类会议,一般是60个工作日,当CDE沟通交流平台当前状态为“待审核”时,即为审核阶段,专业全部反馈完成后,显示为“已反馈”。

3.Pre-INDmeeting到期限后没有反馈可以认为是默许吗?
Pre-INDmeeting是没有到期限后默许的说法的,现阶段由于CDE项目积压,可能会有1~2周的反馈延迟,若到达期限后还未反馈,需要申请人与相关适应症的项目管理人员联系。
4.Pre-INDmeeting到期限后没有反馈,我们可以不等反馈直接提交资料吗?
首先,《化学药品注册受理审查指南》中明确:已申请沟通交流的,应提交与该申请相关的沟通交流编号、沟通交流回复意见,并就回复意见进行逐项答复。
其次,《沟通交流管理办法》中也说明:用于沟通交流的会议资料,应归入申报资料作为审评依据。提交药品注册申请之前的会议重要资料,如《沟通交流会议资料》、《沟通交流会议申请表》、会议纪要等,由申请人归入申报资料一并提交。
因此,除非您对自己的研究及申报资料很自信,可以撤回会议申请提交IND外,还是需要等CDE反馈结束之后才能提交资料。
5.沟通交流会议资料在IND申报资料中如何体现?
沟通交流会议资料提供在申报资料模块一1.6沟通交流会议中。
1.6.1会议申请,可提供会议申请的基本情况,如申请日期、会议编号、会议类型、会议目的等。可以提交沟通交流会议申请表作为附件支持。
1.6.2会议背景资料可参见沟通交流会议申请表。
1.6.3会议相关信函、会议纪要及答复为重点关注内容,可附预约申请单的整个截图,申请人需要对CDE反馈意见进行一一答复。