专家如何看附条件新规(下)|法规观察
转自:研发客
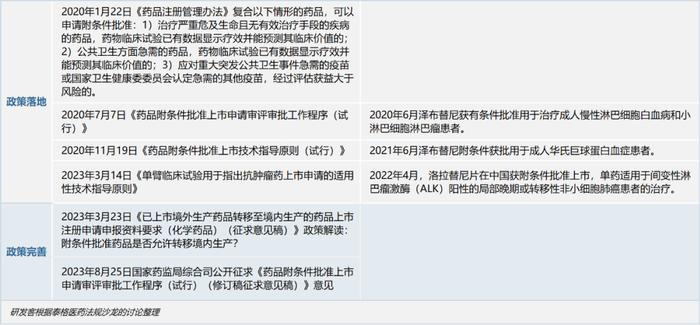
除了附条件批准同类药的数量,确证性研究的完成时限也是《药品附条件批准上市申请审评审批工作程序(试行)(修订稿征求意见稿)》(以下简称《征求意见稿》)的又一讨论热点。
《征求意见稿》明确附条件批准前就要开启确证性研究,4年内完成确证性研究。由此,有不少品种将面临被撤回的境地。作为申请人,如何在相应时间限制内完成附条件上市后确证性研究?在附条件上市申请过程中,与CDE的沟通交流需要注意什么?针对这些产业界关心的问题,研发客继续邀请业内专家各抒己见。
完成确证性研究,4年够吗?
附条件上市申请通过以后,需要开展上市后确证性研究。《征求意见稿》明确要求附条件批准药品在递交申请、与CDE沟通前应启动确证性研究入组,需在4年内完成,明确只有同时满足“治疗领域仍无有效治疗手段、完成入组、综合获益大于风险”情形下才能申请延期,且延期期间药品暂停销售,如不能延期则无法再注册,面临撤市或撤适应症风险。
4年,这个时限对于尚显稚嫩的中国创新药企业而言非常紧迫,据了解,美国FDA附条件批准的药品,从附条件获批到完全获批,最长用时14年。
“附条件批准适用性及上市后要求均趋向于严格,CDE也许主要是考虑临床需求和已有治疗在持续动态变化中,而审评决策取决于目标人群已有的治疗手段。”恒瑞医药全球研发总裁张连山博士说,“相对于现有标准治疗的疗效优势,审评考虑临床研发甚至审评过程会发生动态变化,从而要求创新药企在制定整体临床开发计划(CDP)时要具有前瞻性,及时调整研发策略。”
恒瑞医药注册总监邹莹谈了恒瑞的做法:首先,应尽早与CDE沟通确证性临床方案并达成一致;其次,结合前期研究的数据、进度,入组难度等情况,提前合理规划,选择合适的人群(如前一线),尽早开展上市后确证性研究;最后,研究过程中,持续关注研究入组进度,如有延迟,应及时采取应对措施,如因客观医疗实践发生变化、因科学/伦理等合理原因无法进行,应与CDE及时沟通调整确证性研究方案,以确保研究在法定时限内完成。
不过,研发客在调研中发现,有不少企业早期通过附条件上市的品种因近两年资本寒冬、资金不足等原因难以开展上市后研究。CDE规定附条件批准药品注册证书的有效期不能长于5年。确证性研究数据补充申请的审评时限为200日,因此原则上需要在附条件批准后4年内递交申请。不少品种面临被撤回的境地,这是当下再注册面临的一大难题。
申请延期期间必须暂停销售?
协和麒麟中国研发负责人丁锎博士关注到《征求意见稿》中的“继续研究期间相关药品暂停销售”。
“这一规定对那些没有消极怠工、认真开展承诺性临床试验却遇到现实困难的企业将充满挑战。期待延期期间药品暂停销售的规定具有适度灵活性,可以根据截至延期申请时已获得的临床证据与风险,同时参考企业已开展的临床试验进度。对于是否暂停销售,需要具体品种具体判断。”丁锎说。
对于这一条,勃林格殷格翰注册事务高级总监夏鲲认同丁锎的说法。企业通过附条件批准通道获准上市的项目,应该尽快完成上市后研究,“对于提前上岸的早培生,关键阶段的功课更应该按要求如期完成。”至于未能按时完成作业的特殊情况是否在延期期间停止产品销售,依然要具体问题具体分析,从患者角度出发,看药品对病人的获益程度,以及临床急需性等多方面因素考量。
部分国内企业注册人员也提出“继续研究期间,附条件批准的适应症治疗领域内仍无有效治疗手段,如果该药物评估获益大于风险,药物暂停销售,患者将无药可用。”还有业内人士问到:“如果该药物有多个适应症,部分适应症为附条件批准,在继续研究期间,该如何销售?”
附条件批准的沟通重点在哪
而在沟通交流以及上市后确证性研究等环节,业内并没有太多争议,纷纷表示赞同CDE的《征求意见稿》的专业建议。
以肿瘤药附条件批准的沟通交流为例,主要解决目标适应症所对应的附条件批准适用情形以及何种替代终点或可以被认为是“明显疗效”。美国前FDA高级审评员、思路迪医药首席医学官肖申博士指出,有很多附条件批准的抗肿瘤药的二线和后线治疗的关键性注册研究是通过单臂研究来实现的。今年6月29日,CDE发布了《单臂临床试验用于支持抗肿瘤药上市申请的适用性技术指导原则(征求意见稿)》。
因此,应融合两个指南,制定合理的沟通交流计划。他认为,绝大多数附条件批准药品常用的替代终点是客观缓解率(ORR)的改善。在以ORR作为主要终点指标的单臂设计研究数据中,要通过缓解率合理预测临床获益。
关于《征求意见稿》的沟通交流(II类会议)申请和开展附条件申请批准沟通交流的过程,中国药品监督研究会会长张伟认为,药品审评部门在对药物风险有基本判断、病患者获益大于风险的前提下通过附条件的方式批准药品上市。这也是附条件批准与常规批准沟通交流的不同之处。
恒瑞医药高级注册总监邹莹告诉研发客,在沟通交流过程中,CDE对于疗效获益,通常关注主要终点是否达到预设目标,是否有临床获益,替代终点与临床终点之间是否相关;对于安全性风险,主要关注是否具有严重且无法有效控制安全性风险的不良反应(如不可耐受的肺毒性、心脏毒性等),SAE和死亡事件与研究药物的相关性等,必要时要求申请人进行上市后风险监测评估。
天境生物注册事务部执行总监付洁鹰建议,《征求意见稿》对上市后临床研究相关的沟通交流会议同样能按照II类会议执行,因为上市后研究期限较为紧迫,这样可以留给持有人更多时间开展上市后临床研究。
上市后应严格执行风险管理
《征求意见稿》规定,药品上市许可持有人应当按时限完成附条件研究。对此,中国药品监督管理研究会会长张伟建议企业,要制定好研究计划,定期检查研究进度,发现问题并及时解决问题;有特殊情况尽早与审评部门沟通交流;严格执行风险管理计划,落实好风险控制主体责任;最后,如确实不能按成审评要求完成研究,可尽早主动申请撤销批件。
“就附条件批准上市而言,如果长期不能获得药品的有效性数据,就意味着风险增加,获益减少。这种情形是必须叫停的。”张伟说。
“由于附条件批准一般是基于替代终点、中间指标、早期数据或中期分析甚至是单臂研究,因此,通常研究缺乏长期随访数据。”付洁鹰认为,“上市后研究和风险管理计划应重点关注长期临床疗效确证和安全性风险。”
境内外企业附条件申报经验
截至2022年9月1日,已有32个化学药品或单抗类生物制品的37个肿瘤适应症在境内获附条件批准上市(见下表)。

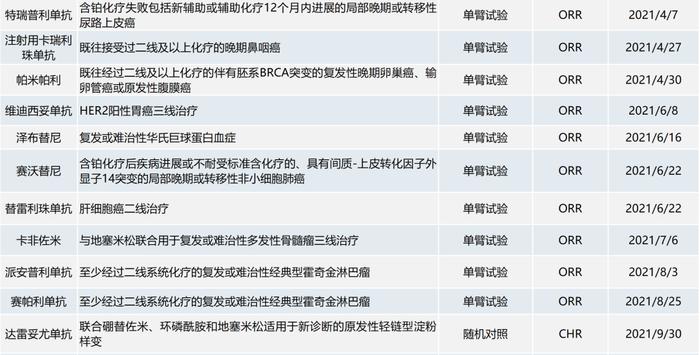
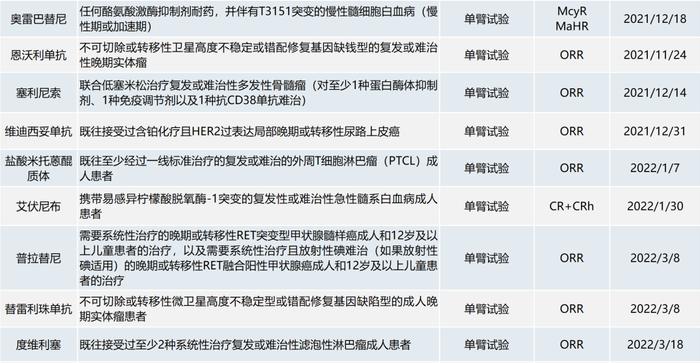
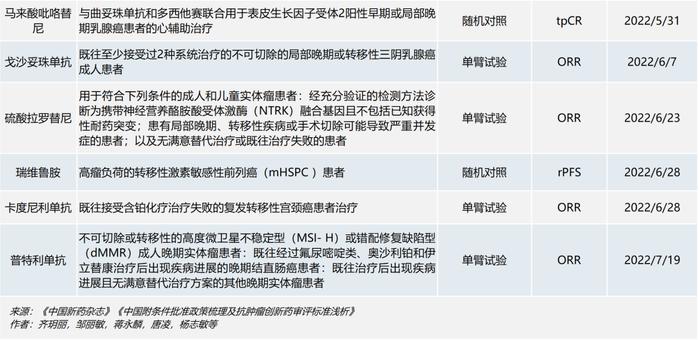
部分境外已上市的药品,因其境外数据显示出突出的治疗获益,中国患者临床需求迫切且存在种族敏感性极小,完全基于境外数据获得“附条件批准”,而上市后须在中国受试者中补充展临床研究。辉瑞和协和麒麟都有在中国附条件获批的经验。
协和麒麟共有3个品种获得附条件批准申请,包括唯一被批准解决X连锁低磷性佝偻病(XLH)和肿瘤性骨软化症(TIO)治疗药物布罗索尤单抗、用于治疗成人中重度斑块状银屑病的布罗利尤单抗以及治疗既往接受过系统性治疗的复发或难治性Sézary综合征或晚期(Ⅲ/Ⅳ)蕈样肉芽肿成人患者的莫格利珠单抗。
据丁锎介绍,布罗索尤单抗最早在欧美日开展多中心试验,出于中国法规政策不明朗,协和麒麟并没有将中国纳入多中心研究。CDE根据药品的临床急需性,布罗索尤单抗在境外获批后,2021年,该药免临床获国家药监局附条件批准上市。后续,CDE要求协和麒麟在中国儿童与成人中开展研究。CDE在承诺性研究的试验设计与病例数要求中考虑了各适应症与人群的流行病学情况与患者入选可行性。
辉瑞中国研发中心总经理陈朝华博士介绍,辉瑞在国内申报上市的产品基本上都是通过参与国际多中心关键3期临床研究与全球同步开发并递交上市申请。有些创新药物在辉瑞总部也属于重点推进甚至称为“光速项目”,在条件满足的情况下,会申请在美国、欧盟等进入快速审评,在国内也会争取附条件批准的通路。
邹莹记得,恒瑞的吡咯替尼最开始是基于一项128例复发或转移性乳腺癌患者的2期临床试验结果,获得了“联合卡培他滨,用于治疗HER2阳性、既往未接受或接受过曲妥珠单抗的复发或转移性乳腺癌患者”的附条件批准,后续顺利完成确证性研究转为“常规批准”。
最后,张伟总结认为,《征求意见稿》是2020年1月22日颁布的《药品注册管理办法》的配套文件之一,旨在总结附条件批准前期工作实践经验,进一步明确标准、规范操作、引导行业理性创新。《征求意见稿》对药品研发行业影响是正面、积极的,起到回归理性研发、避免重复投入、减少内卷和浪费的导向作用。有利于申请人利用有限的临床试验资源,加快药物研究进程,早日批准上市。
接受调研的专家建议,要更多借鉴国外药品审评机构附条件批准的审评标准、考量和策略,开展深入研究,进一步完善《征求意见稿》相关条款,并希望CDE审评多通过培训介绍各适应症领域同类药研发及申报进度,引导行业内良性竞争,并希望附条件批准的药物、适应症以及确证性临床研究的进展等信息可以在网站公示。
“审评部门作为直接的实践者,力争公开透明,最大程度地发挥加快上市注册程序对鼓励创新的激励作用,令患者尽早受益于药品创新的成果。”张伟说。