
2024新书专场 | 《中美欧常用药品注册申报流程汇编》
转自:注册圈

本次书籍由以下企业全程赞助推出:
沈阳悉咨生物医药科技服务有限责任公司
苏州柯里特信息科技有限公司
北京易语堂咨询服务有限责任公司


药品注册是药品研发和上市的关键环节,而中美欧又是全球药品市场的重要区域。对于药企和研发机构而言,了解和掌握中美欧药品注册流程是必备的技能。那么,如何才能全面、系统地了解这些流程呢?《中美欧常用药品注册申报流程汇编》正是您需要的答案之书!
注册圈本次携手10多位行业专家共同整理打造此次书籍。本书籍不仅仅是一个简单的流程汇编,更是一次中美欧药品注册的深度探索。它以实际操作为基础,深入浅出地介绍了中国、美国和欧盟的药品注册流程,为您揭示了那些看似复杂、繁琐的步骤背后的逻辑与规律。
书籍介绍

《中美欧常用药品注册申报流程汇编》这本书从实操的角度,对中国、美国和欧盟的常见药品申报流程和常见问题进行了系统整理。分为三大模块:第一模块为中国部分,涵盖了原辅包登记、临床试验申请、制剂上市申请、一次性进口、沟通交流、再注册、补充申请、变更等申请流程;第二和第三模块为美国和欧盟部分,包括原辅包注册/登记、临床试验申请、制剂注册申请、沟通交流和变更。
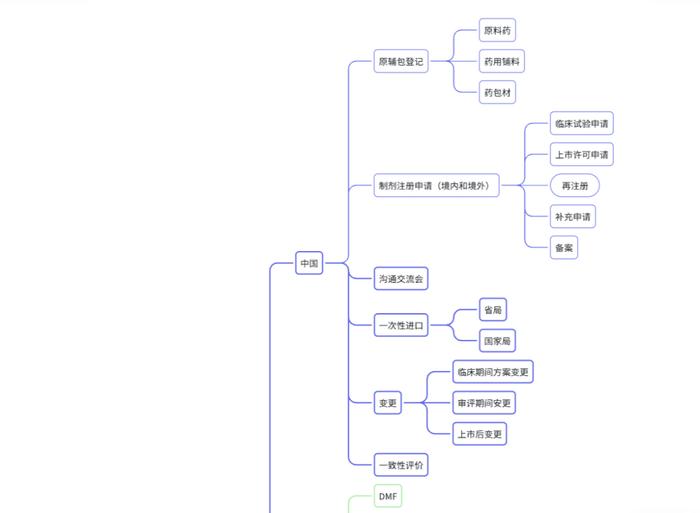
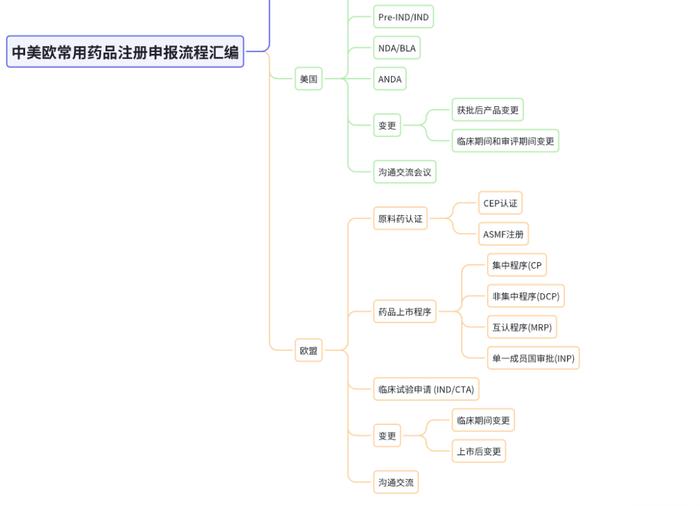
《中美欧常用药品注册申报流程汇编》目录
(下滑查看)
中国药品注册申报流程汇编
1.申报账号创建1
1.1NMPA政务服务门户账号注册1
1.2CDE申请人之窗账号注册3
2. 申报流程6
2.1原辅包登记流程6
2.1.1申报流程图6
2.1.2CDE申请人之窗申报流程7
2.1.3 申报资料清单(NMPA政务服务门户/办事指南)19
2.1.3.1 原料药19
2.1.3.2 药用辅料19
2.1.3.3 药包材20
2.2境内&境外生产药品临床试验申请21
2.2.1 申报流程图21
2.2.2NMPA申报流程22
2.2.2.1 境内生产药品临床试验申请22
2.2.2.2 境外生产药品临床试验申请28
2.2.3申报资料清单35
2.2.3.1 境内生产药品临床试验申请资料清单35
2.2.3.2 境外生产药品临床试验申请资料清单36
2.3 境内&境外生产药品上市许可申请(含新药和仿制药)37
2.3.1 申报流程图37
2.3.2NMPA申报流程38
2.3.2.1 境内生产药品上市许可申请38
2.3.2.2 境外生产药品上市许可申请43
2.3.3 申报资料清单50
2.3.3.1 境内生产药品上市许可申请资料清单50
2.3.3.2 境外生产药品上市许可申请资料清单51
2.4 境内产药品再注册52
2.4.1NMPA再注册申报流程(含药品再注册申报资料清单)52
2.5 境外生产药品再注册申请59
2.5.1 境外药品再注册流程图59
2.5.2NMPA再注册申报流程60
2.5.3 境外生产药品再注册资料列表66
2.6 境内&境外药品补充申请67
2.6.1 申报流程图67
2.6.2NMPA申报流程67
2.6.2.1 境内生产药品补充申请67
2.6.2.2 境外生产药品补充申请75
2.6.3 申报资料列表82
2.6.3.1境内药品补充申请资料清单82
2.6.3.2 境外药品补充申请资料清单83
2.7 境内生产药品备案84
2.7.1NMPA备案流程(含备案资料清单)84
2.8 境外生产药品备案92
2.8.1 备案流程图92
2.8.2NMPA备案流程(含备案资料清单)93
2.9 沟通交流会101
2.9.1 沟通交流会流程图101
2.9.2 沟通交流会分类102
2.9.3CDE申请人之窗申请流程103
2.9.4 沟通交流会资料要求清单106
2.10 一次性进口(国家局&省局)106
2.10.1 申报流程图107
2.10.2NMPA申报流程108
2.10.3 申报资料清单114
2.11 变更115
2.11.1临床期间方案变更115
2.11.2审评期间变更116
2.11.3上市后变更117
2.12 一致性评价118
2.12.1NMPA申报流程118
2.12.1.1 境内生产药品注册一致性评价118
2.12.1.2 境外生产药品注册一致性评价124
3. 电子提交132
3.1电子文件132
3.2资料刻盘135
3.3电子申报资料制作软件135
3.4其他提示136
美国药品注册申报流程汇编
1.美国DMF申报流程138
1.1 美国DMF的介绍138
1.2DMF的类别139
1.3DMF申报的一般要求140
1.4DMF申报流程141
1.4.1DMF号申请141
1.4.2申请ESG账号141
1.4.3 申报资料准备144
1.5DMF增补和年报150
2.美国PreIND和IND申报流程180
2.1美国Pre-IND申请180
2.1.1Pre-IND递交前准备工作180
2.1.2Pre-IND申请流程182
2.2美国IND申请184
2.2.1IND资料准备185
2.2.2IND审评186
2.2.3IND维护 186
2.2.3.1IND的失活、撤回及终止186
2.2.3.2 临床方案增补187
2.2.3.3CMC增补187
2.2.3.4 非临床增补188
2.2.3.5 年报188
2.2.3.6 安全性报告188
3.美国FDANDA/BLA申请操作流程189
3.1 适用范围189
3.2 美国FDANDA/BLA申请前的准备工作189
3.2.1DUNS号(邓白氏号)申请189
3.2.2FEI号(FacilityEstablishmentIdentifier,工厂识别号)申请190
3.2.3ESG(ElectronicSubmissionGateway)账号建立192
3.2.4U.S.Agent193
3.2.5 申请NDA/BLA号193
3.2.6Pre-NDA/BLAmeeting195
3.2.7确认distributor信息以及NDC(NationalDrugCode)号197
3.2.8 专有名称(ProprietaryNames)的申请197
3.3NDA/BLA申报资料的准备199
3.3.1 申报资料序列号199
3.3.2 申报资料主体内容199
3.3.3 申报资料格式200
3.4NDA/BLA申报资料的递交201
3.4.1 申报资料递交前确认201
3.4.2 申报资料的递交202
3.5NDA/BLA审评流程202
3.5.1 计划审评(PlanReview)205
3.5.2 审评(ConductReview)207
3.5.3对申请做出官方决定(TakeOfficialAction)209
3.5.4 审评后的反馈(PostActionFeedback)209
4.美国FDAANDA申请操作流程210
4.1适用范围210
4.2 美国FDAANDA申请前的准备工作210
4.2.1DUNS编码(邓白氏编码)申请210
4.2.2FEI号(FacilityEstablishmentIdentifier,工厂识别号)申请211
4.2.3ESG(ElectronicSubmissionGateway)账号建立212
4.2.4U.S.Agent213
4.2.5 申请ANDA号213
4.2.6 确认distributor信息(如有)以及NDC(NationalDrugCode)号215
4.3ANDA申报资料的准备216
4.3.1 申报资料序列号216
4.3.2申报资料主体内容216
4.3.3 申报资料格式217
4.4ANDA申报资料的递交218
4.4.1 申报资料递交前确认218
4.4.2申报资料的递交221
4.5ANDA审评流程221
4.5.1ANDA接收222
4.5.2ANDA技术审评222
4.5.3 对申请做出官方决定(TakeOfficialAction)224
5.美国获批后产品的变更梳理225
5.1概述225
5.2 变更类别及范畴225
5.3 不同类型的变更评估226
5.3.1 处方与组分变更226
5.3.2 场地变更228
5.3.3 生产工艺变更231
5.3.4 质量标准&分析方法234
5.3.5 容器密闭系统236
5.3.6 标签239
5.3.7 其他类变更241
5.4 变更递交及获批242
5.4.1 非年报类变更资料模板242
5.4.1.1 变更递交资料的模块242
5.4.1.2 变更递交资料的模板243
5.4.2 年报类变更资料模板249
5.5 变更递交流程255
5.6 变更审评流程和实施节点255
5.7 参考指南258
6.美国FDA临床期间变更法规及流程259
6.1 方案修订259
6.1.1范围及类型259
6.1.2递交方式261
6.1.3资料内容及要求261
6.1.4审批及变更的实施261
6.2信息修订262
6.2.1 范围和类型262
6.2.2 递交方式264
6.2.3 资料内容及要求264
6.2.4 审批及变更的实施264
6.3安全报告(safetyreport)265
6.3.1范围和类型265
6.3.2递交方式265
6.3.3资料内容及要求266
6.3.4审批及意见反馈266
6.4 年度报告(AnnualReports)267
6.4.1范围和类型267
6.4.2递交方式268
6.4.3资料内容及要求268
6.4.4审批及意见反馈268
7.美国FDA审评期间变更流程268
8.美国FDA沟通交流会议269
8.1 沟通交流会议类型269
8.2 沟通交流会议的形式269
8.3 沟通交流会议的流程270
8.3.1 申请eCTD的申请编号270
8.3.2 会议申请表(meetingRequest)的撰写和提交271
8.3.3FDA对会议申请的反馈272
8.3.4 会议资料包(MeetingPackage)的递交272
8.3.5FDA会议前的初步回复274
8.3.6会议的召开275
8.3.7 会议的纪要275
欧盟药品注册申报流程汇编
1.欧盟原料药认证276
1.1CEP程序276
1.1.1CEP程序简介276
1.1.1.1CEP网址276
1.1.1.2CEP认证背景276
1.1.1.3CEP互认277
1.1.2CEP程序申请277
1.1.2.1CEP新申请277
1.1.2.2 变更和更新278
1.1.2.2.1 变更278
1.1.2.2.2 更新279
1.1.2.2.3 变更和更新的文件279
1.1.2.3 姐妹档案280
1.1.2.4CEP申请文件的递交280
1.1.2.4.1CESP账户注册281
1.1.2.4.2 递交eCTD文件282
1.1.2.5 评估流程287
1.1.3 技术咨询会议293
1.1.3.1 技术咨询会议293
1.1.3.2 一对一会议294
1.1.4EDQM检查294
1.1.4.1 检查计划294
1.1.4.2检查结果294
1.1.4.3 检查费用295
1.1.4.4检查资源的国际合作和优化296
1.2ASMF注册296
1.2.1ASMF的注册流程298
1.2.2ASMF申报资料要求299
2.欧盟IND/CTA申报流程300
2.1欧盟临床试验申请介绍(ClinicalTrialApplication,IND/CTA)300
2.1.1CTA法规背景300
2.1.2CTIS(ClinicalTrialsInformationSystem)平台300
2.2 临床试验申请流程介绍302
2.2.1 申办程序及时限要求图示302
2.2.2 提交申请305
2.2.3 评估报告-PartI305
2.2.4 评估报告-PartII306
2.2.5 临床试验的决定结果306
2.2.6退出申请307
2.2.7 重新提交申请307
2.2.8增加相关成员国(见“第四点”)307
2.3 临床试验实质性变更程序(Substantialmodification)307
2.3.1 定义307
2.3.2 实质性变更的授权程序307
2.3.2.1 评估报告PartI所涵盖的方面进行重大变更的授权申请验证程序307
2.3.2.2 对评估报告PartI所涵盖的一个方面进行实质性变更评估
308
2.3.2.3 关于对评估报告PartI所涉方面进行实质性变更的决定
310
2.3.2.4 关于对评估报告PartII所涉某一方面进行实质性变更的验
证、评估和决定310
2.3.3 申办程序及时限要求图示313
2.4 后续增加CMS实施临床试验315
2.5 临床试验的实施流程318
2.6 初次申请档案所需材料323
2.7 缩略语326
2.8 参考文献326
2.9Q&A327
3.欧洲药品上市程序328
3.1 欧洲现行的药品法律法规监管体系简介328
3.2 药品上市程序介绍329
3.2.1 集中程序(CentralizedProcedure,CP)329
3.2.2 非集中程序(DecentralizedProcedure,DCP)334
3.2.3 互认程序(MutualRecognitionProcedure,MRP)338
3.2.4 单一成员国审批(IndependentNationalProcedure,INP)340
3.3缩略语340
3.4 参考文献341
4. 欧盟药品临床期间变更342
4.1 临床试验期间药学变更343
4.2EMA关于临床方面实质性/非实质性变更的清单344
5.欧盟药品上市后变更347
5.1 变更程序348
5.1.1IA类微小变更348
5.1.1.1 互认审评348
5.1.1.2 成员国独立审评348
5.1.1.3 集中审评349
5.1.2IB类微小变更349
5.1.2.1互认审评349
5.1.2.2成员国独立审评350
5.1.2.3 集中审评351
5.1.3II类重大变更353
5.1.3.1互认审评353
5.1.3.2成员国独立审评354
5.1.3.3 集中审评355
5.1.4 扩展357
5.1.5紧急安全限制358
5.1.6 关联变更358
5.1.7 其他358
5.2 变更的分类及内容359
5.2.1IA类微小变更359
5.2.2II类重大变更360
5.2.3 扩展360
5.2.4IB类微小变更361
5.2.5 紧急安全限制361
5.3 变更的实施362
5.4 变更提交的资料362
5.5 参考文献363
6. 欧盟沟通交流制度-MAApre-submissioninteractions363
6.1总则363
6.2 早期交互的目的与范围364
6.3 上市许可申请预提交交互的时间安排364
6.4 上市许可申请预提交交互相关人员365
6.5上市许可申请预提交交互需要准备的文件365
6.6 上市许可申请预提交交互的流程366
6.7Pre-submissionmeeting的后续工作367
6.8 与申请评估员的Pre-submissionmeeting367
6.9 上市申请预提交交互表(示例)367
6.10 缩略语374
7.欧盟沟通交流制度流程介绍374
7.1SA&PA申请流程375
7.1.1 在EMA注册375
7.1.2 正式申请和验证申请375
7.1.3 指定专员376
7.1.4 专员召集评估小组并编写报告376
7.1.5 与研发者开会(如果被SAWP要求)376
7.1.6 咨询专家376
7.1.7 咨询患者376
7.1.8EMA最终回应376
7.2 申请程序时间线377
7.3SA/PA费用381
8.2024年科学咨询工作组(SAWP)会议申请递交截止日期-SA,PA,生物标志物鉴定381
9.缩略语382
书籍特色

对中美欧药品注册的常见申请流程进行系统了分类梳理,涵盖的注册类型齐全。
从实操的方式展示了申报和审评的完整流程,包括申报资料要求、资料申报(递交)流程、审评流程(带时间节点)、付费要求,包括账号的创建、注册申请表填写、电子递交、资料刻盘及邮寄等等。
书籍参编人员来源于各大药企和CRO公司,在特定的细分领域具有丰富的理论和实战经验,充分保证了内容的准确性和实用性,是注册人必备的工具书之一。








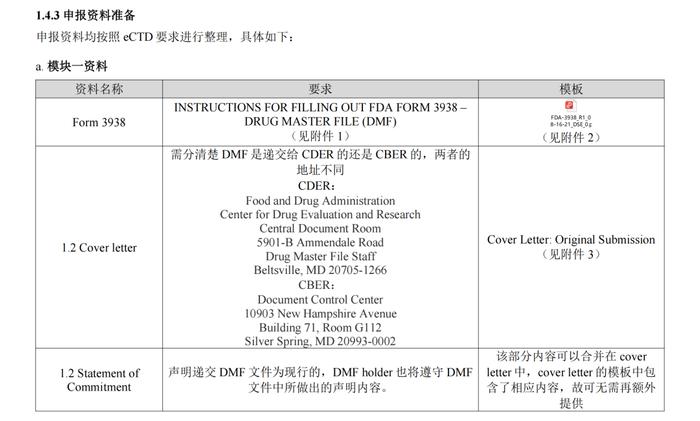
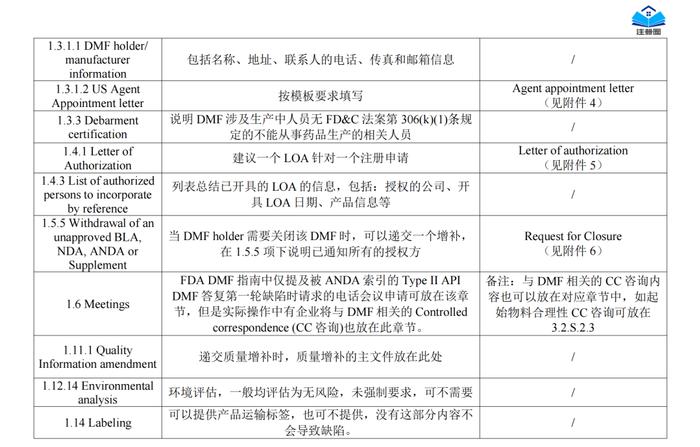

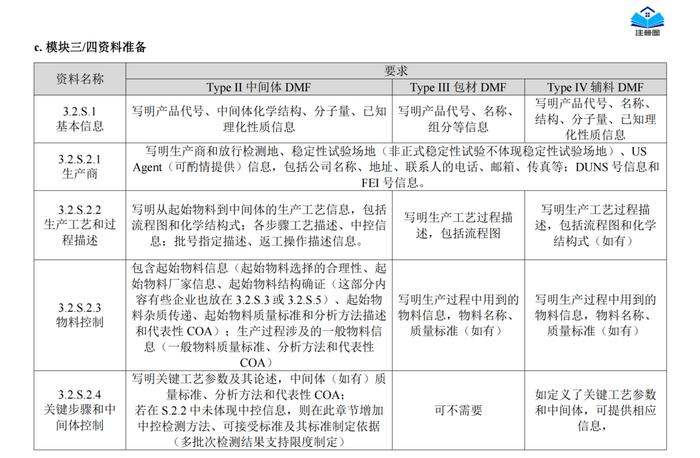
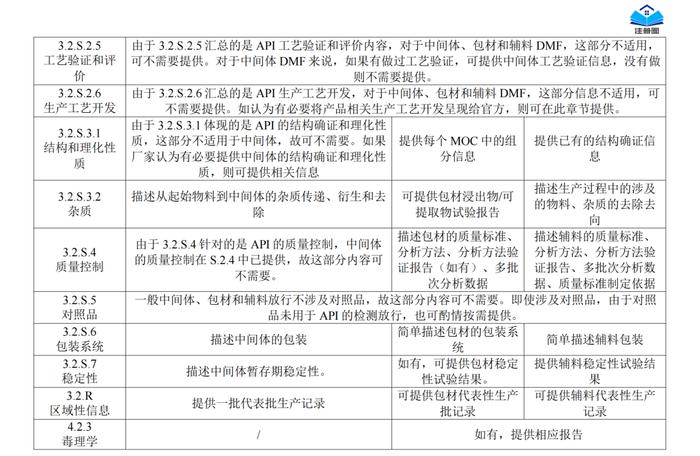


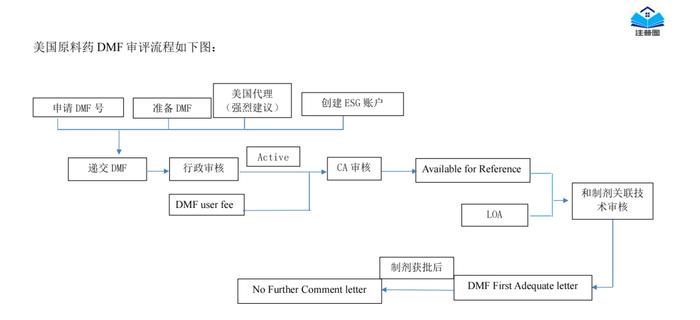
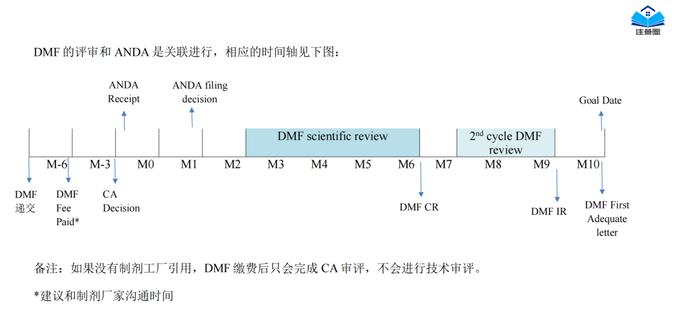
正文精选部分(上下滑动)
书籍评价
为了更进一步保证书籍质量,我们特邀请涉及相关领域专家进行对书籍的评价和建议,供给读者们参考。




新书上架,限时折扣
原价¥249,现在购买,立省¥60
仅需:
¥189元/本
(由于活动价已经低于会员折扣价,故会员在此基础上不再享受八折优惠)

购买网址 ↓↓↓
https://www.regulet.com/
更有超值福利 ↓↓↓
01
满减优惠
●单笔购买满¥1000,立享95折优惠(单本低至¥179.55)
●单笔购买满¥2000,立享9折优惠(单本低至¥170)
● 单笔购买满¥3000,立享85折优惠(单本低至¥160.65)
●单笔购买满¥4000,立享8折优惠(单本低至¥151.2)
02
超值换购
●单笔购买20本及以上,免费赠送书籍*1
●单笔购买50本及以上,免费赠送书籍*1 + 网站199元年会员*1
● 单笔购买80本及以上,免费赠送书籍*1 + DMF英文翻译参考工具书*1+300元注册币返利(可用于购买网站任何产品)
温馨提示:
福利时间:2024年1月17日-2024年3月1日
发货时间:15个工作日(春节期间下单的节后寄送)