
推荐 | 冻干工艺经验谈-国内外仿制药申报思路差异-以注射用培美曲塞二钠为例
转自:注册圈
引言:
由于各种因素,我国医药市场以仿制药为主,自FDA发布包括速释和缓释两种固体制剂的“仿制药QbD实例”进入我国后,国内的仿制药开发纷纷效仿,给我国仿制药开发带来了深远的影响,但是貌似带来的消极影响远比积极影响更多:更多的人选择了这个模板的表象--即模板里有什么我就做什么,无论什么品种什么剂型都在努力的套用这个模板,忽略了不同药物制剂的质量特性差异,也忽略了药物制剂的价值在于满足病患的诉求程度。
正文:
我们说药物制剂的价值在于满足患者诉求的程度,更安全、更有效是药物获得肯定的标准。对于仿制药,特别是在已公布配方组成的前提下,安全性和有效性已经成功了一半,至少我们可以狭义的认为注射剂完全相同的配方是治疗效果完全一致的基石--对“安全”和“有效”排列优先级:使用药物治疗后患者的情况不能变得更差,因此“安全”比“有效”更重要,安全性是首先被考虑的,任何注射剂中细菌内毒素是至热、炎症甚至危及生命严重后果的决定因素,颗粒是导致炎症、血栓的关键因素,而无菌原本是一个相对的概念,即使有微生物存在,不会导致严重危及生命的后果,对于国内更看重的有关物质,除致敏源(比如青霉素本身不会使人过敏,而其杂质使人过敏)外的绝大部分杂质相对于药物活性成分对人的影响小的多,优先顺序从高到低的因素依次是:细菌内毒素、不溶性微粒(包括粒径和粒度分布)、无菌保证值、有关物质。由于个体的差异,一种药物不可能对任何人都起作用,保证更高的起效比率和尽量高的效益/风险比就达到“有效”的目的。
药物制剂从实验室到商业化过程,提供全套的技术解决方案是制剂开发的核心,也是衔接基础研究结果结论到生产力的桥梁,在CMC(ChemicalManufacturingandControl)阶段我们的思路与更高水平国家的思路的差异,我们以注射用培美曲塞二钠为例进行分析。
参比制剂解析
根据“一致性评价”要求,我们对参比制剂进行解析,目的在于获得参比制剂的质量属性信息以设定自制制品关键质量属性。可能根据不同单位的要求,进行含量、有关物质、pH、水分、复溶时间、不溶性微粒、可见异物等等的检测,然后是影响因素的检测,通过这些实实在在的看得见摸得到的数据来为自己的实验设计建立基础。我个人认为这种宏观的方式不一定是好的方法,药物制剂的化学降解来源于组分间的相互作用的因素微乎其微,因为除药物活性成分外的其他辅料一定是为了药物活性成分在制剂形式中更稳定而存在的。对于制剂的理化性质,在给定的质量标准或进口注册质量标准中已经完全明确了基本的质量属性,前期对参比制剂的解析意义难道只是为了印证参比制剂满足质量标准要求?
参比制剂的解析,更多的是透过表象看本质,如何看到质量标准背后那个原研制剂的内控标准,做到与参比制剂具有相同质量属性的自制品需要采用哪些除公布的质量标准项下外的衡量标准。这要求我们看到微观的层面:根据药物制剂的给药方式、代谢特点及药物活性成分的化学稳定性确认自制品的关键质量属性并建立相关的质量标准。
培美曲塞二钠七水物结果如下:
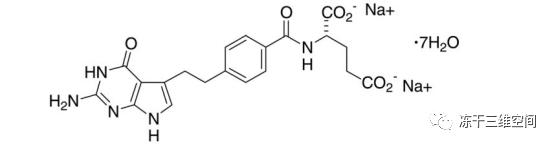
两种规格制剂分别为100mg和500mg(以培美曲塞计),100mg规格制品含有106mg甘露醇,500mg规格制品含有500mg甘露醇,用氢氧化钠或盐酸调pH至7—8。该药物静脉注射给药、配方具有良好的水溶性,注定该制品在安全性考量上有且仅有细菌内毒素、不溶性微粒、无菌保证值三项,有效性考量只要药物活性成分是培美曲塞、剂量足够这一项。
试验设计
首先,实验室小试阶段的处方考察,既然原研药已经证明了其配方是化学稳定性满足质量要求的,我们还需要纠结考察培美曲塞二钠的化学性质以及制剂不同pH条件、不同辅料用料条件下的制剂稳定性么?我认为大可不必:原研药足够的安全性试验结果已经足够了,甚至已经清清楚楚的写在说明书里,对于配方的变化已经没有考察的意义。从这个角度来说,既然已经掌握了配方的性质,在进行物料的风险评估过程中,全部因素都是低风险。
其次,实验室小试阶段的工艺考察,两种水溶性极好的物料溶解在水中,且两种物料均未接近饱和溶解度,最终在溶解的过程中只剩下温度及作用时间对酰胺水解程度的影响,考察不同温度条件下有关物质的增长对时间的函数就完成了。在灭菌程序的选择上无需多言,采用干热、湿热及辐射灭菌都无法实现,只能采用除菌过滤的形式。在滤器材质的选择上恰恰是我们做的欠缺的地方:更多的考察滤器材质的吸附性、相容性,实验室无法提供恒压条件下药液过滤速度结果和恒流条件下滤器压力升高直至最大安全压力结果,因此在这个阶段只需要采用证明水性滤器材质不会在药液作用下产生脱落物或破损就可以了。对于细菌内毒素和不溶性微粒,这个阶段没有条件进行考察。对于冻干,配方中添加大量的甘露醇、且不同规格制品甘露醇用量稍有差异,可能说明药物活性成分是一种质轻的成分,关注升华速率与漂粉的关系也足够了,至于获得一条什么样的冻干曲线,我个人认为试验冻干机的捕冰效率和传热效率与商业化冻干机相差极大,没有必要确认具体的冻干曲线,当然,能够通过传热系数和升华阻力计算不同冻干机上大致冻干周期时,试验冻干机的冻干参数无需考虑是否满载,更不需要大量的冻干试验。
如下图是使用50R西林瓶装载20mL散装药液的500mg规格(以培美曲塞计)制品的状态,制品存放的时间很长了,但是我们能够看到瓶内壁上均匀附着的粉末,培美曲塞的冻干过程一定要控制好一次干燥初中期的升华速率,在可接受范围内最大限度降低漂粉情况。


再次,从实验室到商业化的转化过程中,当药液配制批量增加,需要确认合理的散装药液配制方式和工艺参数,需要证明散装药液在配制过程中与直接接触药液的罐体、管道等材质的相容性,需要根据工艺过程中的无菌保证水平以及药物活性成分化学性质设计适当的试验证明工艺设计的合理、准确性,需要确认除菌过滤器与药液的相容性,还需要设计试验证明根据包装材料选择依据选择的包装材料的准确性,这个过程是一个不断完善的验证过程,最终通过多批次的验证生产证明工艺能够生产出质量稳定的制品。实际上从实验室到商业化的转化过程往往是我们所欠缺的。
综上,国内申报与国外申报的差异:国内更看重的是实验室开发过程按照指导原则没有缺项,对于工艺的稳定性评估是欠缺的;国外更看重工艺的稳定性,对于实验室小试,并经不是GMP条件下的商业化过程,即使一笔带过都没有问题。国外更注重GMP条件下的商业化活动赋予制剂的批间质量属性差异以及生产管理水平,以评价生产的制剂的实际质量水平和生产单位保证生产符合要求制剂的能力。
申报资料的侧重点差异
国内的申报资料要求不缺项,国外的申报资料要求依据,国内的申报资料重点放在了制品的开发历程中,国外的申报资料重点在于强调制品商业化生产的质量属性和证明制品质量属性的依据以及药物制剂临床使用过程中接触其它容器后对患者的安全性。
国内现有的QbD模版出现在申报资料“产品开发”中,国外申报资料中“summary”部分恰恰将开发历程简化到配方说明和工艺描述程度,一些趋势对比更是简化到用曲线图的方式表述,更多的篇幅用于工艺验证部分的内容以证明工艺可控性。置于QbD,是质量控制白皮书中的内容,目的是更好的引检和更充分的表述制品的质量说明及依据和开发思路,QbD完全不是“产品开发”或者“summary”部分要表述的。
注射用培美曲塞二钠的细节,在后续文中更新,欢迎交流心得。