
在这迷人又壮美的科学领域,"中国玩家"能奋起直追吗?

► 文 观察者网专栏作者 潘禺
2016年3月,在观看了AlphaGo在围棋这一古老游戏中击败了人类世界冠军李世石后,DeepMind联合创始人德米斯·哈萨比斯回想起了自己本科时期的经历。
他当时玩过一个名为Foldit的游戏,玩家可以在游戏中将氨基酸链折叠成蛋白质结构,哪怕玩家对生物学一无所知,并不影响他们折叠蛋白质。如果DeepMind能用AI来模仿围棋大师的直觉,难道不能编写一个算法,用AI来模仿Foldit玩家的直觉吗?
蛋白质折叠问题
蛋白质折叠是一个迷人的问题。
一张纸,在没有折叠前,不过是压扁的木浆。当你折叠这张纸,就能产生各种功能。比如折成飞机,那么纸飞机就能被投掷并滑行,供孩子们娱乐。而如果折成灯笼,就能在中秋节赏玩,表达团圆的美好寓意。
地球上已知的蛋白质,是拥有数亿种不同形状的分子,每一种都执行特定的生物学功能。血红蛋白和肌红蛋白在肌肉和身体中运输氧气,角蛋白赋予头发、指甲和皮肤结构,胰岛素使葡萄糖进入细胞转化为能量。这些功能,通常由蛋白质的形状或结构定义。一串氨基酸分子,在没有自发折叠成其固有形状之前,就没有功能。
一个细胞将称为氨基酸的小分子串联成多肽链,这就是制造蛋白质的过程。细胞如何选择氨基酸,取决于DNA提供的底层指令集。多肽链一旦组装好,在极短的时间,千分之一秒内,会弯曲、再弯曲,精确地折叠成蛋白质的最终三维形状,随后离开分子装配线,立即去执行它的生物学工作。
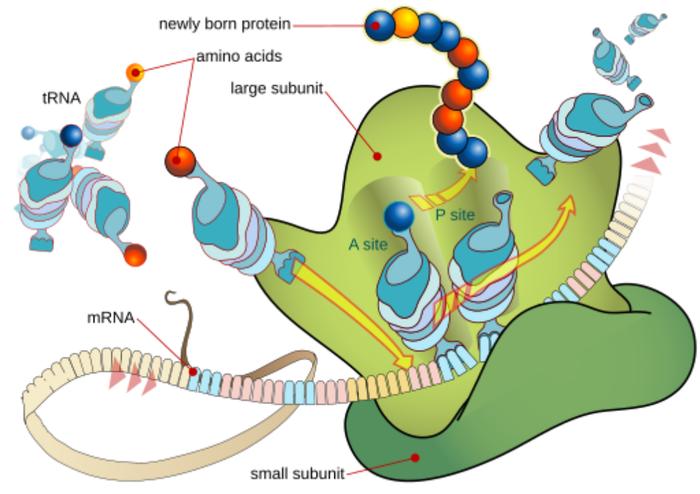
如果蛋白质执行这种折叠过程出了差错,错误折叠或解缠,就可能导致毒性和细胞死亡。许多疾病,如镰状细胞性贫血,就是由错误折叠的蛋白质引起的。错误折叠的蛋白质聚集成团,是阿尔茨海默病和帕金森病等神经退行性疾病的标志。
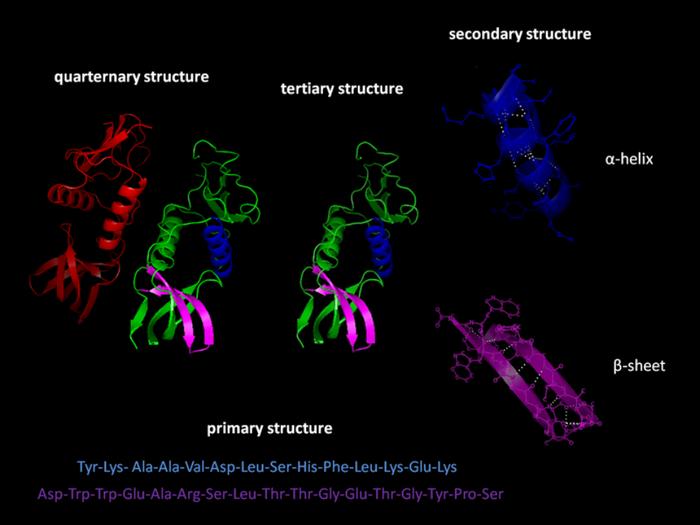
已知的蛋白质结构可以分为四个层次。
一级结构可以理解为一条线性的字符串。基本组成单元是一个个的氨基酸,即一个个的字母。常见的氨基酸只有20种,所以一级结构的字符串通常只包含20种字母,不包含的6种字母是BJOUXZ。二级结构就是在一级结构的字符串的基础上,肽链进行折叠变换,形成一种局部的三维结构。三级结构就是把多个二级结构拼接到一起,折叠成一个完整的蛋白质三维结构。四级结构就是多个三级结构分子组合成一个复合物。
20世纪50年代,生物化学家克里斯蒂安·安芬森的发现使他获得了诺贝尔奖。他将蛋白质添加到化学溶液中,溶液的破坏导致蛋白质错误折叠,但他接下来观察到,去除化学剂后,蛋白质还是可以自发地重新折叠,恢复其天然的结构。安芬森假设,蛋白质折叠成其原始结构是由蛋白质的氨基酸序列自动完成的,氨基酸序列里就包含了所需的全部信息。这就是安芬森教条。
安芬森教条意味着,应该有一种方法可以从氨基酸序列预测蛋白质的形状,这就是蛋白质折叠问题。
分子生物学中的许多假设被称为教条(dogma),最著名的是中心法则(Thecentraldogmaofmolecularbiology),遗传信息的标准流程是DNA制造RNA,RNA制造蛋白质,中心法则指出,遗传信息传到蛋白质后,不会回流到核酸之中。蛋白质折叠领域还有一个教条,叫莱文塔尔(Levinthal)悖论,说的是一个给定的蛋白质可供选择的可能构象的数量是天文数字,即使是一个小蛋白质,也需要比宇宙存在的时间更多的时间来探索所有可能的构象,可谓“一沙一世界,一花一天堂”。
安芬森教条的例外,则是人类已知的许多疾病。比如朊病毒的构象,就与应有的原生折叠状态不同。淀粉样蛋白疾病,如牛海绵状脑病(疯牛病)、阿尔茨海默病和帕金森病,都是安芬森教条的例外,原生蛋白错误折叠成不同的构象,从而导致致命的淀粉样蛋白堆积。
回到蛋白质折叠问题,蛋白质组装的时间这么短,到底是什么东西,将蛋白质引向正确的折叠路径呢?能否从氨基酸序列预测蛋白质的结构?折叠的代码和机制是什么?
为了搞清楚这些问题,至少必须先用实验确定蛋白质的结构。科学家将蛋白质培育成晶体,用X射线轰击它们,并测量射线的弯曲,这就是X射线晶体学。20世纪60年代,生物学家马克斯·佩鲁茨和约翰·肯德鲁用这种方法确定了血红蛋白和肌红蛋白的3D结构,又一项获得诺贝尔奖的工作产生了。
随着更多蛋白质结构被发现,科学家们在1971年建立了蛋白质结构的免费档案库——蛋白质数据银行。最初,只包含了七种蛋白质的结构。近50年后,谷歌DeepMind使用它来训练AlphaFold时,已经包含了超过140000种。
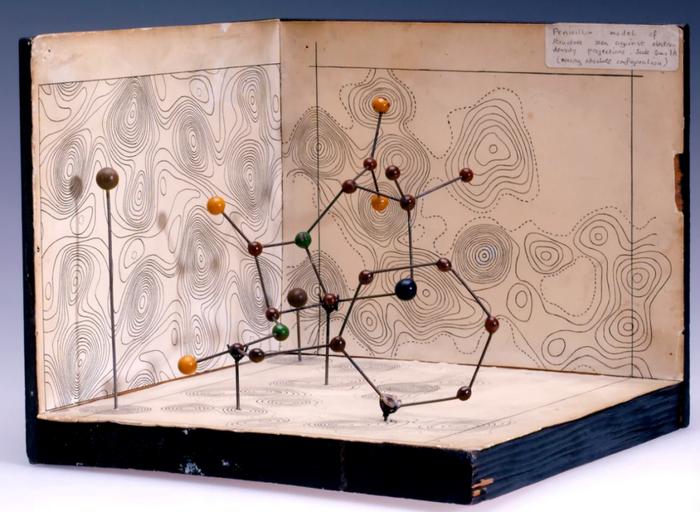
因为方法的繁琐,为蛋白质银行添砖加瓦的过程,曾经是非常艰难痛苦的。科学家们先要创建蛋白质电子密度图,在电子聚集的区域可能包含一个原子。将电子密度图打印到塑料片上,一个个堆叠起来,就创建了蛋白质地理的“等高线图”。然后,科学家们要将地图转换为物理模型,将塑料地图放入理查兹盒中,这个设备以发明者牛津大学生物物理学家理查兹的名字命名,在理查兹盒内,一定角度的镜子将地图反射到工作区,使科学家能准确看到每个原子的相对位置。然后,科学家们就用球和棍子物理构建他们的模型。
为了研究并模拟磷酸化酶,科学家不得不爬上梯子进入一个两层楼高,特别建造的理查兹盒中。这种蛋白质拥有842个氨基酸,是当时人们研究过的最大的蛋白质。由于进展的煎熬和缓慢,蛋白质银行成立的20年后,有信心确定而被提交的蛋白质结构也不过七百多种。
实验主义与计算主义
主张计算方法的科学家,已经厌倦了实验派的做法,他们希望另辟蹊径。
正如安芬森的教条,蛋白质的结构应该能从其氨基酸序列中预测出来。计算生物学家编写计算机算法,希望可以给程序输入一串氨基酸,生成正确的蛋白质结构。对计算方法来说,蛋白质的三维结构预测问题,可以看成这样一个问题,输入是一个字符串,输出是每个字符(残基)对应的三个扭转角ϕ、ψ和ω,看起来简洁漂亮。注意,这看起来和AI处理的一些经典问题,如序列标注、机器翻译等问题很像。
计算派的做法是在虚拟世界构建自己的模型,设计自己的算法,比如假定原子以某种方式粘在一起,蛋白质总是这样向右或向左折叠,但这些模型逐渐远离现实。
实验主义者工作精确但速度慢;计算主义者工作迅速,但与生物物理现实脱节,常常出错。两种方法的优点,必须结合起来。实验派和计算派的科学家,必须牵手合作。
物理学家普朗克有过一句名言:“一个新的科学真理的成功,并不是因为它征服了那些反对者并使他们顿悟,它的成功是因为它的那些反对者最终逝去,而心向新理论的新生代最终成长起来。”
普朗克说的应该是科学理论,是有哲学高度的理论解释。或许正因为理论还难以建立,在蛋白质生物学的发展历程中,我们看到的并不是这样残酷的规律,而是反对派之间的合作共进。在20世纪90年代,科学家们组成了社区,实验主义者提供最新的蛋白质氨基酸序列清单,计算主义者则尽其所能,用他们想要的任何方法来预测蛋白质的结构。一个独立的科学家小组,通过将计算派的模型与实验确认的结构进行比较,来评估模型。
这个名为CASP的社区,成了解决蛋白质折叠问题各种计算方法的试验场,最后实际上已经变成了一场竞赛。在美国加州的一座老教堂里,计算主义者可以在会议中谈论他们的方法,组织者鼓励与会者,如果不喜欢他们听到的内容就在木地板上跺脚。据一位生物学家回忆:“一开始,有很多跺脚,几乎就像打鼓一样。”
一些方法的表现比预期好,比如“同源建模”,比较已知蛋白质的结构来推断未知蛋白质的结构。其他的则完全没有用。在1998年的比赛中,大卫·贝克用他的算法罗塞塔(Rosetta)大放异彩,罗塞塔算法模拟了氨基酸分子间原子的相互作用,以预测它们将如何折叠。尽管还不够准确,无法实用,但人们看到了计算预测蛋白质结构的曙光。
2008年,贝克创建了一个名为Foldit的免费在线电脑游戏,也就是本文开头所说的那个游戏。在当时,人类玩家模拟蛋白质超过了罗塞塔,但人类的领先优势不会持续太久。
如果两个氨基酸一起突变,它们可能有某种联系,可能在空间上很接近,这一概念被称为共同进化。在清除了统计方法引入的错误后,科学家提高了对哪些氨基酸共同进化的预测准确度,基于此,罗塞塔算法能更准确预测蛋白质结构,这可能是深度学习之前推动该领域进步的最大里程碑之一。但共同进化需要大量相似的蛋白质进行比较,而实验主义者解析蛋白质结构的速度不足以满足计算主义者的需求。
新玩家上场
2016年,谷歌DeepMind的人工智能团队以深度学习算法在围棋中击败了人类冠军,轰动了世界。
深度学习本身就是计算机科学受到生物学启发的范例。在大脑皮层中,分子信息被发送到神经元相互连接的网络中。神经元有叫做突触的小臂,它们抓住邻近神经元发出的分子,这些分子告诉接收神经元要么发射并传播信号,要么不发射。
将电子位连接起来创建“神经网络”的想法,早在20世纪50年代就已经在计算机科学中产生。神经网络中的每个单元是一个节点,可以比作神经元:一个神经元从其他神经元接收信息,然后计算是否向接下来的神经元发射。在神经网络中,信息在多层神经元中传播,以产生特定的结果,比如图像识别。神经元层数越多,可以执行的计算就越复杂。
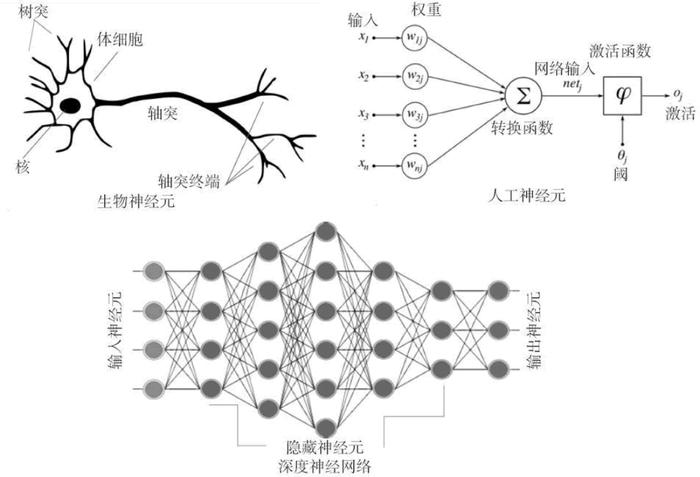
这一灵感正是来自大脑。神经科学发现,我们的大脑会通过逐步抽象的方式来分析眼睛所看到的事物。在AI应用中,输入数据的传感器可以是镜头、麦克风或者其他测量仪器。而我们人类眼睛中的传感器又被称为视锥细胞和视杆细胞,它们会探测那些令其进入激发状态的光线,得到光线的亮度和颜色。这相当于计算机图像中每一个像素的亮度和颜色。人类的第二层神经元会连接着眼睛的视锥细胞和视杆细胞,一般会衡量相邻像素之间的相关度,根据上一层神经元的激活情况来计算。下一层神经元可以在眼睛看到的图像中找出明显的线条,再下一层,会将线条结合起来,得知图像中的基本对象,比如绵羊的耳朵。再之后的层次,继而将这些基本对象结合起来,确定更深层次的结构,比如图像中是否存在绵羊。
2010年代初,计算机科学家已经能更好构建神经网络,允许更多层的可靠训练。网络深度从之前的两三层,跃升到数千层。为了区分过去浅层的做法,人们开始用“深度学习”这个更时髦的名字来称呼。深度学习改变了人工智能,算法不仅在图像和声音的识别上表现出色,在围棋这样的游戏中也能击败人类。近年来,基于深度学习的自然语言处理模型GPT,则在文本生成上又一次震撼了世界。
这里多说几句题外话,当前的人工智能革命,还与一种概率论思想——贝叶斯方法(Bayesianmethods)有关。贝叶斯方法的核心思想是根据观测数据更新先验概率,得到后验概率分布。贝叶斯方法将不确定性视为概率分布,能够量化模型的不确定性。在深度学习中,许多问题涉及到对不确定性的建模,例如参数估计、预测的置信度等。用贝叶斯方法,能够更加灵活地处理这些问题。
一些科学家甚至相信,我们的大脑就是一个能对贝叶斯公式进行各种各样近似计算的计算器,也就是贝叶斯大脑,贝叶斯公式很可能在人类认知中处于核心位置。贝叶斯主义者的信念也深刻影响了当前人工智能的发展。总之,“生物学太重要了,不能只留给生物学家”,为了努力理解不同的蛋白质如何折叠,人们不仅要研究生物,还要研究数学、物理、化学、统计学、计算机科学……
百图生科首席AI科学家宋乐在谈到其大模型时就说过:“不单单需要AI人才,也有工程人才的参与,此外还需要一些很了解生物知识、对生物数据分析很有经验的人才。这种团队的内部合作不容易,但如果成功也会收效颇丰。”
随着谷歌DeepMind进入蛋白质结构预测领域,受生物学启发的深度学习,现在要来解决生物学中的难题了。
AlphaFold的小小震撼
DeepMind的这个项目称为AlphaFold,来自统计学、结构生物学、计算化学、软件工程等领域的专家,在DeepMind共同研究蛋白质折叠问题。在学术界,专家们通常相互隔离,各自独立进行项目,很少有这样的合作,更没有谷歌庞大的财务和计算资源支持。2017年,蛋白质数据银行已经拥有超过140000种结构,DeepMind团队用这些数据训练他们的算法。
其领导者约翰·贾姆珀(JohnJumper)正是在物理、化学、生物学和计算机方面有着多样化的背景。贾姆珀从小自学了编程,本科学习数学和物理,先攻读凝聚态物理学博士,后来退学在纽约的一家公司用超级计算机从事蛋白质的模拟,通过理解蛋白质的运动和变化,希望更好地理解各种疾病,如肺癌的机制。此后又在芝加哥大学学习理论化学,完成了博士学位。

2018年春天,AlphaFold已经准备好参加CASP,人工智能要与真正的蛋白质科学家竞争了。CASP组织者最终带来的消息是,AlphaFold表现得非常好,在预测蛋白质结构方面,比第二名的团队好大约2.5倍。但这离解决蛋白质折叠问题还很远。
在贾姆珀的领导下,AlphaFold被更新重建了,DeepMind设计了一种新型的Transformer架构,神经网络调整了其连接的强度,以创建更准确的蛋白质进化和结构数据表示。
AlphaFold2的预测效率和准确性有了巨大提高。DeepMind找了大约50篇发表在《科学》、《自然》和《细胞》等高端期刊上的论文,这些论文都是实验主义者的辛勤工作成果,描述新的蛋白质结构和功能,将AlphaFold2的预测结果与之对照,可以继续打磨改进。
在2020年的CASP比赛中,评估员将预测的蛋白质结构与经过验证的实验结构进行比较来打分,100分即模型和现实在原子层面上完全匹配。AlphaFold2的大多数结构都达到或超过了90分。大多数情况下,算法都有效。
DeepMind已经解决了蛋白质折叠问题中的结构预测部分。AlphaFold2能够准确地根据其氨基酸序列预测蛋白质的结构。对于因疫情封锁在家,通过Zoom参加CASP会议看到AlphaFold2演示的科学家们来说,这个小小震撼意味着,蛋白质科学的世界已经永远改变了。
设计蛋白质:逆蛋白质折叠问题
长期以来,实验生物学家对计算持怀疑态度,AlphaFold2的成功无疑改变了这一点,但如果说“改变一切”,就有些夸大其词。
AlphaFold2并不等于结构生物学家的失业。
当然,失业总会存在。一些细胞生物学家和生物化学家过去常常与结构生物学家合作,现在他们已经用AlphaFold2来取代。尽管,训练AlphaFold的数据,是结构生物学家过去用一个个实验精心确定的。
结构生物学的技术,除了前面说的X射线晶体学,还有冷冻电镜、NMR波谱、双偏振干涉测量等技术。
而中国公众可能对冷冻电镜比较熟悉。这一昂贵的设备(Cryo-EM,冷冻电子显微镜),其原理是快速冷冻生物样品并用电子束轰击它们。X射线晶体学需要蛋白质结晶,而冷冻电镜能够处理非晶态样品。X射线晶体学在高分辨率原子级结构方面有优势,而冷冻电镜在解析大型复合物和动态过程中更为强大。过去十年中,冷冻电镜发展迅速,成为解析复杂生物大分子结构的重要工具之一。广为公众熟知的中国科学家施一公、颜宁等人,都是用冷冻电镜解析蛋白质结构的专家。

如果结构生物学家仅仅研究蛋白质结构,那他们当然失业了。但结构生物学家的目标是发现蛋白质的功能。有了AlphaFold2,他们就有了一个更好的工具,可以在几分钟内创建一个假设,而不是等待几个月甚至几年通过实验来确定一个结构。
结构生物学家的角色不仅仅是获取结构数据,还包括解释这些数据、设计实验验证假设,并理解蛋白质功能和与疾病相关的机制。这个问题就像AIGC会不会让创作者失业。ChatGPT能告诉你的答案也许准确度已经很高,但未必完美,对AI大模型生成的内容,每一个创作者都还需要仔细甄别、验证,并理解这些内容的真正意义,用这些内容为自己和社会创造价值。
AlphaFold2的不完美在于,在预测简单的小型蛋白质结构方面,非常出色,但在预测包含多个部分的蛋白质,动态蛋白质(与其他分子相互作用时,形状会发生变化)时,准确性较低。有时,蛋白质需要被特定的离子、盐或金属包围才能正确折叠,自然环境会改变蛋白质的形状,AlphaFold2并不能考虑。
仅仅识别已知蛋白质的结构和功能是不够的。
对于新药研发来说,科学家需要设计那些在自然界中不存在的蛋白质,这就是蛋白质设计,也可以理解为“逆蛋白质折叠问题”。还记得用AI解决蛋白质折叠问题是什么意思吗?无非就是向深度学习算法输入氨基酸序列,要求其输出蛋白质结构。这个问题逆过来,就是设计师将一个蛋白质结构输入算法,并要求其输出氨基酸序列。然后,设计师使用那个氨基酸序列在实验室中构建蛋白质。
宋乐就曾讲过,要设计一段有效的蛋白质,“有20个不同的位置,每个位置有20种不同的选择。这是一个巨大的空间,人的思维很难对这个空间进行整体的筛选或对比,而计算机来做这件事就有巨大优势。”
Foldit游戏的创建者贝克,就做了一个专门用于设计的算法,称为RoseTTAFolddiffusion,Foldit游戏本身也更新了设计蛋白质的版本。蛋白质设计并非新鲜事物,但深度学习加速了其发展。以前,训练有素的蛋白质设计师需要花费数周或数月的时间,才能创建新蛋白质的主链。现在他们可以在几天内,甚至一夜之间完成。
AlphaFold3与中国玩家
2022年,谷歌DeepMind发布了全球已知的2.18亿种蛋白质的结构预测,这几乎就是所有。其竞争对手Meta公司也于当年推出了蛋白质结构预测模型ESMFold。
但AlphaFold2仍有缺陷,比如前文提到的无法考虑环境。细胞内部是复杂的生物学环境,充满了各种分子——蛋白质、信号分子、信使RNA、细胞器等,蛋白质不是独立工作,而是不断与其他分子相互作用,这会改变其自身的形式和功能。将细胞分子的景观渲染出来,可视化呈现,你会看到非常壮美的复杂性。

AlphaFold2的能力限于预测单一蛋白质结构,而要帮助生物学家理解这个复杂原生环境中的蛋白质,就是这一领域现在的发展方向。2024年春,谷歌DeepMind更新算法,发表了AlphaFold3的论文,大卫·贝克则推出了RoseTTAFoldAll-Atom算法,都致力于能够预测蛋白质互相结合,或与DNA、RNA和其他小分子结合时的结构。
AlphaFold3能预测分子复合物的结构,比如某种在植物真菌中发现的酶。根据业内专家的分析,目前这些算法的准确性仍有待改进,不太可能很快带来新药。一个重要变化是,AlphaFold2的基础代码是开源的,每个人都可以研究算法并为自己的项目重建,但谷歌没有开源AlphaFold3。
中国企业也在加入AI+生命科学的领域。基于AlphaFold2算法改进,华为昇思MindSpore团队,采用自己的昇腾计算平台,在2022年4月一度拿下CAMEO这一蛋白质结构预测竞赛的第一名,这个比赛每周都会在线更新分数和名次。
2020年创立的百图生科,则致力于搭建“xTrimo”生命科学大模型,这是一个雄心勃勃的超大规模多模态模型体系,在底座通用模型上,除了蛋白质生成模型,还有多个下游任务模型共同组成。比如靶点发现,也就是免疫细胞扰动后功能变化预测模型。
当发现了一个疾病靶点后,就要设计一个蛋白质。
如果将疾病相关的靶点想象成一把锁,设计蛋白质就是配钥匙,要打开锁,锁齿和钥匙就要契合,这就需要模型来预测。因此蛋白质生成不仅要预测结构,还要预测蛋白质与靶点的契合度,也就是结合的紧密强弱,然后再对AI生成的许多设计做筛选,将最合适的送去试验。
xTrimo有多个层次,第一层是对单个蛋白质的建模,第二层是对细胞中蛋白质相互作用的建模,第三层是对细胞本身的建模,第四层则是对细胞系统的建模。因而,这个体系不仅能表征单体蛋白质,还能表征蛋白质相互作用、免疫细胞、免疫系统等多层次生物问题,帮助研究者更快发现新的蛋白质、新的细胞形态,发现新的靶点和药物设计方向。
为此,百图生科构建了世界最大的免疫图谱,包含66亿个蛋白,超300亿条蛋白互作关系,1亿个单细胞,以及超6100万条免疫互作关系和6000亿条泛细胞共现关系。
结语
生命体的高度复杂,还远不是AI科学家使用的庞大但依然有限的数据量就能揭示的。
蛋白质折叠问题仍未完全解决。AI能识别出给定氨基酸序列可能的折叠模式,但蛋白质折叠过程中,究竟发生了什么,其中的信息依然是黑箱。对于理解整件事发生的过程,AI并不能给出答案,深度学习算法无法告诉我们基于蛋白质的生命机制和本质,无法告诉我们背后的基本物理原理。如果只有结果,没有过程,这还是科学吗?
无论如何,科学确实在前进。70年前,人们还认为蛋白质只是一种凝胶状物质。但今天,我们看到了蛋白质世界的一个又一个结构。